Role and regulation of prolyl hydroxylase domain proteins (original) (raw)
- Review
- Published: 15 February 2008
Cell Death & Differentiation volume 15, pages 635–641 (2008)Cite this article
- 41k Accesses
- 352 Citations
- 4 Altmetric
- Metrics details
Abstract
Oxygen-dependent hydroxylation of hypoxia-inducible factor (HIF)-α subunits by prolyl hydroxylase domain (PHD) proteins signals their polyubiquitination and proteasomal degradation, and plays a critical role in regulating HIF abundance and oxygen homeostasis. While oxygen concentration plays a major role in determining the efficiency of PHD-catalyzed hydroxylation reactions, many other environmental and intracellular factors also significantly modulate PHD activities. In addition, PHDs may also employ hydroxylase-independent mechanisms to modify HIF activity. Interestingly, while PHDs regulate HIF-α protein stability, PHD2 and PHD3 themselves are subject to feedback upregulation by HIFs. Functionally, different PHD isoforms may differentially contribute to specific pathophysiological processes, including angiogenesis, erythropoiesis, tumorigenesis, and cell growth, differentiation and survival. Because of diverse roles of PHDs in many different processes, loss of PHD expression or function triggers multi-faceted pathophysiological changes as has been shown in mice lacking different PHD isoforms. Future investigations are needed to explore in vivo specificity of PHDs over different HIF-α subunits and differential roles of PHD isoforms in different biological processes.
Similar content being viewed by others
Main
In response to hypoxia, metazoans from worms to humans increase glycolysis to compensate for energy loss due to reduced oxidative phosphorylation, and upregulate erythropoiesis and angiogenesis to achieve more efficient oxygen utilization. Central to this adaptive response is the mechanism that uses oxygen deficiency as a signal to activate the transcription of genes important for these processes. The key to this activation mechanism is the rapid accumulation of hypoxia-inducible factor (HIF)-α under hypoxia.1, 2 In essentially all cell types examined so far, HIF-α subunits undergo oxygen-dependent hydroxylation on specific proline residues. Hydroxylated HIF-α subunits are recognized by the von Hippel–Lindau (VHL) protein of the E3 ubiquitin ligase complex, and are rapidly destroyed via the polyubiquitination/proteasomal degradation pathway.3, 4, 5, 6, 7 As intracellular O2 concentration is reduced, non-hydroxylated HIF-α subunits accumulate and form functional transcription factors in the nucleus by heterodimerization with the constitutively expressed HIF-1_β_subunit.1
HIF-prolyl hydroxylases (HPHs),6 also referred to as prolyl hydroxylase domain (PHD) proteins,7 form an evolutionarily conserved subfamily of dioxygenases that uses oxygen and 2-oxoglutarate (2-OG) as co-substrates, and iron and ascorbate as cofactors.6, 7, 8 While these enzymes are most intensively studied in animals especially mammalian cells, a similar hydroxylase has also been described in the plant Dictyostelium, where it hydroxylates SKP1, a homolog of the FBOX component of the E3 ubiquitin ligase.9 In addition, enzymes with similar catalytic centers may also exist in some prokaryotes.10 While Drosophila and Caenorhabditis elegans each has a single family member, known as dHPH in Drosophila6 and _eg_g _l_aying _n_ine (EGLN or EGL-9) in C. elegans (for its essential egg-laying function),7 mammals have four members belonging to this subfamily, including PHD1/EGLN2/HPH3, PHD2/EGLN1/HPH2, PHD3/EGLN3/HPH1 and a recently characterized protein named as P4H-TM.6, 7, 11 Several other names have also been used for mammalian homologs, including SM-20 for rat PHD37 and HIF-P4H1, HIF-P4H2 and HIF-P4H3 to indicate that hydroxylation occurs on position 4 of proline residues.12 Given that the term ‘PHD proteins’ both reflects the functionality as hydroxylases and avoids committing them as being HIF specific (because non-HIF substrates also exist13), we will refer to mammalian homologs as PHD1, PHD2 and PHD3, however, keeping the name P4H-TM11 for the fourth member of the subfamily due to the fact that other names have not been used in the literature.
While O2 concentration is the most ubiquitous factor controlling PHD-catalyzed hydroxylation reactions, there is now strong evidence that various other parameters may also come into play.14 Furthermore, hydroxylation of non-HIF substrates and hydroxylase-independent functions have also been reported for PHDs or their plant homologs.9, 13, 15, 16 Thus, in addition to serving as oxygen sensors, PHDs may potentially have other functions as well. In this review, we provide a summary and analysis of current literature on PHDs and discuss their roles in regulating oxygen homeostasis as well as other pathophysiological processes.
Regulation of PHD-Catalyzed Hydroxylation Reactions
Both full-length PHD1 and PHD2 have over 400 (407 and 426 in humans, respectively) amino-acid residues and share a well-conserved hydroxylase domain at their C-terminal halves (∼55% identity within hydroxylase domains), whereas the N-terminal halves are more divergent and have poorly characterized functions.7 The much shorter PHD3 (239 amino-acid residues in humans) contains the hydroxylase domain but only a short stretch of the divergent N-terminal sequence. Since basic enzymatic mechanisms of PHD hydroxylases have been reviewed extensively,2, 8, 17 here we focus on how the hydroxylase activity is regulated (Figure 1).
Figure 1
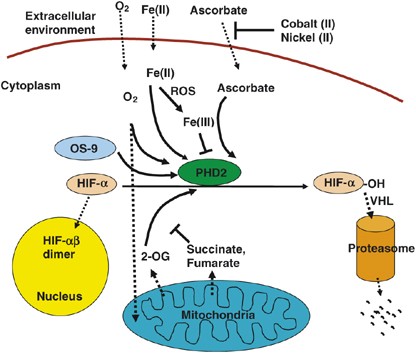
Schematic illustration of regulatory mechanisms of PHD hydroxylase activities. Regulation of hydroxylase activity by small molecules and protein factors are indicated, but regulation at the level of PHD expression is not shown. Dashed arrows indicate movements of molecules in the direction of the arrows, but solid arrows indicate functions in favor of the indicated direction. For example, ascorbate is indicated by dashed arrow from the extracellular environment into the cytoplasm to show that it is imported from outside, but the solid arrow between ascorbate and PHD2, following the direction of HIF-1_α_ to HIF-1_α_-OH reaction, indicates that ascorbate supports this reaction. On the other hand, a ‘T’-shaped symbol indicates the interference of certain functions. An example of this is the interference of the role of 2-OG by succinate and fumarate
Oxygen. Oxygen is a key substrate for hydroxylation. All three PHDs have _K_m values for oxygen within the range of 230–250 _μ_M, slightly above the oxygen concentration in aqueous solutions saturated by ambient room air.12 Since intracellular oxygen concentrations are typically below these values, high _K_m values ensure the dependence of hydroxylase activity on oxygen when all other substrates and cofactors are present at adequate levels.
Cells cultured under severe hypoxia (0.5–2% oxygen) almost invariably accumulate HIF-1_α_; however, recent studies found that such accumulation is dependent on functional mitochondria.14, 18 Respiratory inhibitors, such as nitric oxide (NO) which inactivates cytochrome c oxidase, causes destabilization of HIF-1_α_ even under hypoxia.14, 18 One theory proposes that functional mitochondria rapidly consumes oxygen through oxidative phosphorylation, thus diminishing O2 concentration in the cytosol18 (Figure 1). When mitochondrial oxygen consumption is inhibited, oxygen is ‘redistributed’, resulting in an increase in cytosolic oxygen concentration. Under normoxia, however, cytosolic oxygen levels may remain sufficiently high for active hydroxylation in spite of rapid consumption by mitochondria. This interpretation is consistent with the finding that mitochondrial inhibitors do not substantially increase PHD activities under normoxia.14
In addition to oxygen-dependent prolyl hydroxylation mediated by PHDs, HIF-1_α_ is subject to oxygen-dependent asparaginyl hydroxylation by factor inhibiting HIF (FIH).19, 20 FIH is also an iron-dependent dioxygenase and hydroxylates Asn 851 located within the C-terminal transactivation domain of HIF-1_α_. Hydroxylation at Asn 851 inactivates HIF-1_α_ by preventing its interaction with CBP/p300, an essential coactivator for HIF-dependent transcription.19, 20
Intracellular Fe(II) concentration. NO may inhibit PHD activity by chelating Fe(II) or other mechanisms in addition to the previously mentioned role in promoting PHD activity by oxygen redistribution21, 22 (Figure 2). The net outcome may depend on which effect happens to dominate and is more easily observed under specific experimental conditions. For example, while high concentrations of NO (>1 μ_M) inhibit PHD activity, the resultant stabilization of HIF-1_α is easily detectable only under normoxia, whereas under hypoxia such an effect is masked by hypoxia-induced HIF-α accumulation. Lower NO concentrations (<0.4 _μ_M) are insufficient to inhibit PHD activities, thus the ability to promote PHD activity (and hence destabilizing HIF-α) plays a major role. However, such a role is prominent only under hypoxia, whereas under normoxia the contribution of NO-induced increase in PHD activity may be negligible compared to PHD activation by high concentrations of oxygen.18
Figure 2
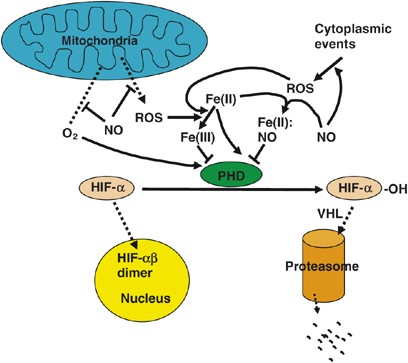
Regulation of PHD hydroxylase activities by NO and ROS. These molecules may have multiple points of interaction with PHD regulatory mechanisms or originate from different sources, which is consistent with the complexity of the effects of NO and ROS revealed in various studies. Merging of arrows originating from Fe(II) and NO indicate binding between Fe(II) and NO; other symbols are the same as explained in Figure 1
Reactive oxygen species (ROS) may inhibit PHDs by chelating and oxidizing PHD bound Fe(II) to Fe(III)14, 23 (Figure 2). Mitochondrial ROS production is associated with oxidative phosphorylation, but interestingly ROS production increases with reduced availability of oxygen due to stress.24, 25 Thus, oxidative phosphorylation under hypoxia may inhibit PHD activity by abundant ROS production.14 In this regard, NO or other mitochondria inhibitors may activate PHD activity by preventing ROS production in mitochondria,14 a mechanism that is not mutually exclusive with the oxygen sink theory18 (Figure 2).
Of note, one study found that defective oxidative phosphorylation under hypoxia (hence less ROS) did not compromise hypoxia-induced HIF-1_α_ stabilization,26 suggesting that ROS effects on PHD activity are potentially complicated. On the other hand, ROS is not only generated in mitochondria but also by various cytoplasmic activities (Figure 2). For example, JunD_−/− mutation leads to elevated RAS signaling and ROS production, which leads to decreased PHD activities and HIF-1_α stabilization.23 Under conditions where cytosolic ROS production is significantly elevated, a reduction in mitochondrial ROS production may not have much noticeable effect on total ROS concentration and therefore fails to further inhibit PHD activity. This interpretation may help explaining some discrepancies in the literature.
Some environmental factors, such as nickel (II) and cobalt (II), can inhibit PHD activity and cause HIF-α stabilization.27 Interestingly, exposure of cells to these ions greatly reduced intracellular ascorbate concentrations. However, PHD activities could be restored by supplying higher concentrations of ascorbate to culture media, suggesting that nickel (II) and cobalt (II) block the entry of ascorbate into the cell27 (Figure 1). These findings suggest the importance of ascorbate in PHD activities, likely by reducing PHD-bound Fe(III) back to Fe(II).
Competitive inhibition by 2-OG analogs. 2-Oxoglutarate, an intermediate of the tricarboxylic acid (TCA) cycle, is an essential co-substrate for PHDs due to its role in Fe(II) coordination in the catalytic center.7 However, mitochondria also generate inhibitory TCA cycle intermediates, including succinate and fumarate which compete against 2-OG for binding to the active site28 (Figure 1). Under normal circumstances, these molecules probably do not significantly interfere with PHD function due to the fact that their cytosolic concentrations are very low. However, mitochondrial defects or manipulations that lead to significant cytosolic accumulation of these intermediates are associated with abundant HIF-1_α_ accumulation due to competitive inhibition of PHD activities.14, 28, 29, 30 Another TCA cycle intermediate oxaloacetate and the glycolysis product pyruvate were also reported to inhibit PHDs in some experimental systems but not in others, suggesting that their effects may be complicated and are influenced significantly by experimental conditions.14, 31
Prolyl substrate specificity. Hydroxylation occurs on the fourth position on P402 and P564 in human HIF-1_α_ (or at similar positions in HIF-2_α_) within the so-called oxygen-dependent degradation domains (ODDs).32 The ODD domain was originally identified as a region containing about 200 amino-acid residues that overlap with the transactivation domain;32 however, subsequent studies have shown that two independent subregions, each containing an LXXLAP core with short flanking sequences, were hydroxylated at the proline residue of the LXXLAP core.33 While both proline residues are located within the C-terminal half of HIF-α, each LXXLAP core along with its short flanking sequences is termed N-ODD or C-ODD, respectively, depending on their relative proximity to the N and C termini.
All PHDs are more active on the C-ODD than N-ODD, and in particular PHD3 is virtually inactive on N-ODD.12 In addition, PHD2 hydroxylates HIF-2_α_ N-ODD less efficiently than HIF-1_α_ N-ODD.12, 34 Detailed mechanisms of ODD selectivity are not known. In PHD2, however, several antiparallel _β_-sheets, formed by the sequence approximately between positions 197 and 380, are likely involved in ODD interaction. Patches of hydrophobic amino-acid residues in this region are likely the most important elements in mediating ODD substrate recognition.10
Surprisingly, besides the proline residue itself, other conserved residues of the LXXLAP core are not essential for substrate recognition.35, 36 Gal4-ODD fusion proteins carrying various mutations in the core sequence (except for the proline residue itself) were hydroxylated almost as efficiently as wild-type controls.35 However, mutations beyond the conserved core significantly affected hydroxylation efficiency.37 Interestingly, when positions of N-ODD and C-ODD were exchanged, the efficiencies of their hydroxylation by PHD1 and PHD2 were also changed accordingly. Nevertheless, PHD3 remained exclusively active on the original C-ODD even after it was moved to the N-ODD position.38
In cultured cells, all three PHDs contribute to the regulation of both HIF-1_α_ and HIF-2_α_, but preferences do exist. PHD1 and PHD3 are more active on HIF-2_α_ than HIF-1_α_, whereas PHD2 hydroxylates HIF-1_α_ more efficiently.34 Consistent with these cell culture-based studies, PHD2 deficiency in mouse liver and kidney tissues was associated with the accumulation of nuclear HIF-1_α_ but not HIF-2_α.39 In contrast, PHD1/PHD3 double deficiency led to hepatic accumulation of HIF-2_α but not HIF-1_α.40 Although relative abundance of different PHDs in a particular cell type may contribute to differential hydroxylation, preferences still exist even in cells expressing comparable levels of different PHDs. Furthermore, even though PHD3 is the most efficient in C-ODD hydroxylation, it does not compensate for the loss of PHD2 in destabilizing HIF-1_α.34 At the meantime, the lack of PHD3 hydroxylase activity toward the N-ODD does not explain why it cannot replace PHD2 because N-ODD hydroxylation contributes only minimally to HIF-1_α_ degradation.37
These complexities indicate that selectivity of HIF-α hydroxylation by different PHDs may go beyond ODD sequence motifs or even hydroxylase activity alone. In this regard, it is important to note that other proteins do exist that modify hydroxylation activity by means of protein–protein interactions. For example, OS-9 brings HIF-1_α_ and PHDs into a multi-protein complex and promotes hydroxylation.41
Regulation of PHD Availability
PHD2 was initially found to be the most critical hydroxylase in a large variety of cultured cells;42 however, subsequent analyses found that one important reason was its more abundant expression than other isoforms.34 When PHD1 and PHD3 expression levels were increased by transfection, the importance of these isoforms was also revealed, especially in the hydroxylation of HIF-2_α._34 These studies illustrate that regulation of PHD abundance plays a crucial role in oxygen sensing.
Tissue and intracellular distribution. At the RNA level, all three _PHD_s are widely distributed among different organs, although PHD1 is highest in testes whereas PHD3 is the highest in the heart.43 At the protein level, however, PHD2 is the most abundant in all mouse organs examined,40 which is consistent with other studies demonstrating PHD2 being the most abundant isoform in different cell lines.34, 42
Intracellular localization patterns of three PHDs have been studied. While initial analysis using PHD-GFP fusion proteins indicated differential intracellular localization of different PHDs,44 a subsequent study using monoclonal antibody indicated that all endogenous PHDs were mostly located in the cytoplasm.45 Interestingly, P4H-TM, which is related to PHDs, is anchored in the ER membrane with its catalytic domain located in the lumen rather than cytosol.11 In spite of such an orientation, the enzyme hydroxylates HIF-1_α_ and triggers HIF-1_α_ degradation.
Transcriptional regulation. PHD2 and PHD3 genes contain hypoxia response elements46, 47 and are induced by HIF-1_α_ under hypoxia46, 48, 49, 50 (Figure 3). Not surprisingly, inhibition of PHDs by NO or other molecules may also result in feedback upregulation of PHDs due to HIF-α accumulation.51 Besides serving as a negative feedback mechanism under hypoxia, HIF-induced PHD expression may also ensure rapid removal of HIF-α after reoxygenation.49, 52 Feedback loops may exist at different levels of hypoxia signaling. For example, while PHD2 expression is induced by hypoxia, TGF-_β_1, which is also induced by hypoxia, inhibits PHD2 transcription through Smad-mediated signaling.53 Thus, TGF-_β_1 seems to counter the effect of HIF-induced upregulation of PHD2.
Figure 3
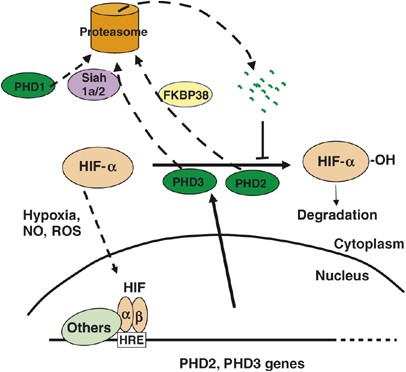
Regulation of PHD expression and abundance. PHD2 and PHD3 genes contain hypoxia response element (HRE) and are transcriptionally upregulated by HIF. Hypoxia or inhibitors that prevent HIF-α degradation promote HIF-α nuclear translocation and heterodimerization with HIF-1_β_. This feedback upregulation results in increased PHD2 and PHD3 abundance, which suppresses further accumulation of HIF-α. PHDs are also subject to proteasomal degradation, assisted by Siah1a/2 or FKBP38 as indicated
Regulation of stability. While the major function of PHDs is to regulate the stability of HIF-α, PHD1 and PHD3 themselves are targeted for polyubiquitination and proteasome-mediated degradation by Siah1_α_/254 (Figure 3). Interestingly, Siah2 itself is strongly induced by hypoxia, which contributes to accelerated PHD degradation, especially that of PHD3.54 Thus, while PHD3 transcription is induced by hypoxia, its protein degradation is also accelerated.54 Even though Siah1_α_/2 do not regulate PHD2 stability, FKBP38, a peptidyl prolyl cis-trans isomerase, plays a moderate role in destabilizing PHD2 (Figure 3). Interaction between the N-termini of FKBP38 and PHD2 mediates PHD2 destabilization but the process is independent of isomerase activity.55
Hydroxylase-Independent or HIF-Independent Functions
PHDs may also inhibit HIF-1_α_ transcriptional activity in addition to reducing its abundance. A complex formed between PHD2 and ING4, a tumor repressor protein, inhibits HIF-α_-mediated expression of angiogenic cytokines and consequently suppresses tumor angiogenesis15 (Figure 4). In support of a hydroxylase-independent role, we also found that overexpression of hydroxylase-deficient PHD2 mutant diminished the stimulating effect of hypoxia on the proliferation of cultured endothelial cells, without altering HIF-1_α abundance.16 In another study, overexpression of PHD2 in a VHL-deficient cell line also inhibited HIF-1_α_ activity although HIF-1_α_ abundance remained the same.52
Figure 4
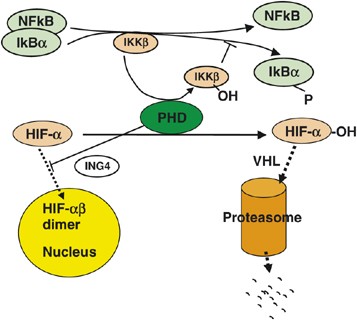
PHDs employ multiple mechanisms to regulate oxygen homeostasis, including regulation of HIF-α abundance, activity and NFκB activity. -P and -OH indicate phosphorylation and hydroxylation modifications, respectively. Dashed lines with arrows at the end indicate molecular movements. Labeled ovals promote a process if placed along a solid line with arrow; blunting with a short perpendicular line at the end indicates inactivity or negative regulation
Map organizer 1 (Morg1), a WD-repeat protein, serves as a scaffold protein for the interaction between HIF-1_α_ and the divergent N-terminal sequence of PHD3.56 This interaction reduces HIF-1_α_ target gene expression without decreasing HIF-1_α_ abundance,56 suggesting possible involvement of a hydroxylase-independent mechanism. However, it is also likely that formation of a complex between Morg1, PHD3 and HIF-1_α_ might instead prevent hydroxylated HIF-1_α_ from entering the degradation pathway.
Hydroxylase-dependent but HIF-α_-independent functions have also been described for PHD1 and PHD2 in regulating the transcriptional activity of NFκB13 (Figure 4). The non-HIF hydroxylation substrate involved is IKK_β, a kinase that in its non-hydroxylated form phosphorylates inhibitory factor IκB_α_. Phosphorylated IκB_α_ dissociates from NFκB, resulting in the activation of NFκB transcriptional activity. Under normoxia, IKK_β_ is hydroxylated by PHD1 or PHD2, and such a modification inhibits its kinase activity. Thus, hydroxylated IKK_β_ fails to trigger dissociation of the inhibitory IκB_α_ from NFκB. When PHD1 or PHD2 is knocked down by siRNA, or inhibited by hypoxia, IKK_β_ hydroxylation is inhibited, thus allowing its kinase activity to phosphorylate IκB_α_, causing its dissociation from NFκB.
Biological Functions of PHDs
Various functions have been found for different PHDs, which are summarized in Figure 5. In C. elegans, loss of EGL-9, the prototype of PHDs, is associated with significantly elevated levels of HIF-α and loss of egg-laying function.7 In Drosophila, its single PHD, termed dHPH,6 hydroxylates Similar (HIF-1_α_ homolog) and causes its oxygen-dependent degradation.57, 58 Drosophila PHD is essential for cell growth in fat bodies and wing imaginal discs.58, 59 In humans, heterozygous P317R mutation in PHD2 is associated with moderate polycythemia.60 P317 is only two amino-acid residues downstream from D315, which directly coordinates Fe(II),7, 10 so P317R mutation likely reduces hydroxylase activity by altering Fe(II) and substrate binding. In addition, R371H mutation in human PHD2 is associated with erythrocytosis, due to reduced binding to, and therefore hydroxylation of, HIF-1_α._61
Figure 5
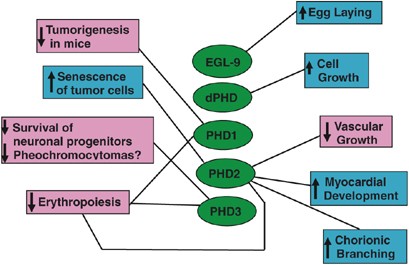
Currently identified biological functions of PHDs. In vivo processes regulated by PHDs are indicated in text boxes, with upward arrows indicating promotion of a process and downward arrows indicate suppression of a process
PHDs are also implicated to function as tumor suppressors. For example, ectopic overexpression of PHD1 suppresses HIF-1_α_ accumulation and tumor growth,62 and immortality of human endometrial cancer cells is often associated with loss of PHD2, while re-introduction of PHD2 induced senescence.63 Likewise, PHD3 is also a potential tumor suppressor. During normal development, many precursors of sympathetic neurons undergo apoptosis, for which PHD3 is uniquely essential.64 Loss of PHD3 is associated with failure of apoptosis and development of pheochromocytomas.64 Interestingly, overexpression of PHD3 (but not PHD1 and PHD2) in PC12 cells promoted apoptosis upon NGF withdrawal.64 Although PHD3 hydroxylase activity is required for this function, expression of mutant HIF-α subunits that cannot be hydroxylated did not prevent PHD3-mediated apoptosis, suggesting that a novel hydroxylase substrate may be involved.
Besides PHD3, PHD2 may also promote cell death, but in a different cell type. Following ischemia–reperfusion treatment of murine heart explants, large infarcts are typically formed due to extensive death of cardiac cells. PHD2 knockdown significantly reduced infarct sizes in ischemia–reperfusion-treated hearts, and such a protective effect may be mediated by HIF-1_α_ stabilization and HIF-mediated expression of iNOS.65 However, PHDs may have apparently opposite effects in different cell types. In maturing chondrocytes, for example, PHDs promote survival rather than apoptosis. Dimethyloxalylglycine (DMOG), which inhibits all three PHDs, sensitized chondrocytes to H2O2-induced apoptosis, whereas HIF-1_α_ knock down by siRNA conferred resistance to H2O2.66
PHDs may also regulate cell differentiation. For instance, C2C12 skeletal myoblast differentiation is accompanied by increased expression of PHD3, and differentiation is blocked by siRNA-mediated knock down of PHD3.67 Interestingly, PHD3 directly binds to myogenin and protects it from VHL-dependent polyubiquitination. Thus, HIF-α hydroxylation is not directly involved in this process, although it remains to be determined if PHD3 directly hydroxylates myogenin. In another example, PHDs were shown to be important for the maintenance of osteoblast identity, because inhibition of PHD activities with DMOG caused osteoblasts to adopt adipogenic phenotypes.68
We have recently investigated roles of PHDs in mice. While _Phd1_−/− or Phd3_−/− mice survive normally, Phd2_−/− embryos die at mid-gestation stages.69 Major defects include underdeveloped myocardium and trabeculae in the heart, and poor labyrinthine branching in the placenta, but embryonic vascularization was apparently normal. Interestingly, significantly increased HIF-1_α, HIF-2_α and VEGF-A levels were detected in the embryo proper but not specifically in the heart, suggesting either the involvement of a systemic effect of PHD2 deficiency or a HIF-independent role in heart development. In adults, tamoxifen-induced, Cre-_loxP_-mediated Phd2 knockout in a broad range of tissues led to increased vascular density and lumen sizes, accompanied by efficient recruitment of vascular smooth muscle cells.39
Surprisingly, even though Phd2 knockout essentially failed in the brain, likely due to blood–brain barrier to tamoxifen, vascular overgrowth occurred similarly as in other tissues.39 Furthermore, increased VEGF-A was found in the serum but not in local tissues where vascular overgrowth was present, and other important angiogenic molecules such as angiopoietin-1 and VEGF receptor-2, were not significantly increased. These findings may be explained by proposing systemic effect where circulating angiogenic factors such as VEGF-A and erythropoietin (EPO, which is also increased) might have potential contribution to increased vascular growth.39
PHD2-deficient adult mice overexpressed EPO by ∼230-fold and developed severe polycythemia.40 Interestingly, besides an expansion of Ter119+ erythropoietic progenitor cell population, liver- and spleen-derived Lin− Sca-1+ CD117+ cell population (enriched in hematopoietic stem cells) was also greatly expanded, which had increased hematopoietic activity in vitro. Thus, PHD2 deficiency might modify microenvironments in the liver and spleen and activate extramedullary hematopoiesis. A cautionary note, however, is that due to the chronic nature of PHD2 deficiency, it remains to be defined if increased extramedullary hematopoiesis is a primary or secondary defect. Adult Phd1_−/−/Phd3_−/− double knockout mice also displayed polycythemia, although milder than in PHD2-deficient mice.40 Surprisingly, renal and serum EPO levels decreased significantly; however, hepatic EPO expression and erythropoietic progenitor numbers did increase. In contrast to PHD2-deficient adult mice where only increased HIF-1_α_ was detected in nuclear extracts, Phd1_−/−/Phd3_−/− double knockout mice displayed increased HIF-2_α_ but not HIF-1_α_ levels in the liver, which is consistent with cell culture studies, indicating that PHD1 and PHD3 were more efficient on HIF-2_α._34
Concluding Remarks
The major driving force behind the intensive interest in prolyl hydroxylases is their potential to serve as therapeutic targets to treat various diseases. One prospective application is to promote angiogenesis in ischemic tissues, for which proof of principle has been demonstrated in mice by inhibiting HIF-α hydroxylation using small molecule hydroxylase inhibitors70 or competitive peptides containing HIF-ODD sequences.71 Other therapeutic applications are also promising. For example, a recent study reported that TM6008, a novel inhibitor that binds to PHD active site and chelates Fe(II), reduced neuronal cell death in a model of cerebrovascular disease without affecting angiogenesis.72 Also, FG-2216, an orally available PHD inhibitor, effectively and reversibly promotes erythropoiesis in rhesus macaques by inducing EPO expression.73 FG-2216 is now in clinical development to treat anemia (http://www.fibrogen.com/trials/). While these findings are all encouraging, it should be cautioned that PHDs are also potential tumor suppressors and it remains to be determined if long-term exposure to PHD inhibitors may favor tumor development. Nonetheless, given the presence of three PHDs, and now the discovery of a fourth related hydroxylase, P4H-TM,11 it may be possible to exploit differential tissue expression profiles and HIF-α specificity to achieve the best therapeutic outcomes while minimizing associated side effects.
Abbreviations
2-OG:
2-oxoglutarate
DMOG:
dimethyloxalylglycine
EGLN:
egg laying nine
HIF:
hypoxia-inducible factor
HPH:
HIF-prolyl hydroxylase
ODD:
oxygen-dependent degradation domain
PHD:
prolyl hydroxylase domain
ROS:
reactive oxygen species
TCA:
tricarboxylic acid
References
- Wang GL, Jiang BH, Rue EA, Semenza GL . Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92: 5510–5514.
Article CAS Google Scholar - Kaelin WG . Proline hydroxylation and gene expression. Annu Rev Biochem 2005; 74: 115–128.
Article CAS Google Scholar - Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271–275.
Article CAS Google Scholar - Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464–468.
Article CAS Google Scholar - Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ et al. Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292: 468–472.
Article CAS Google Scholar - Bruick RK, McKnight SL . A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001; 294: 1337–1340.
Article CAS Google Scholar - Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR et al. C.elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001; 107: 43–54.
Article CAS Google Scholar - Schofield CJ, Ratcliffe PJ . Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun 2005; 338: 617–626.
Article CAS Google Scholar - van der Wel H, Ercan A, West CM . The Skp1 prolyl hydroxylase from Dictyostelium is related to the hypoxia-inducible factor-alpha class of animal prolyl 4-hydroxylases. J Biol Chem 2005; 280: 14645–14655.
Article CAS Google Scholar - McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM et al. Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci USA 2006; 103: 9814–9819.
Article CAS Google Scholar - Koivunen P, Tiainen P, Hyvarinen J, Williams KE, Sormunen R, Klaus SJ et al. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem 2007; 282: 30544–30552.
Article CAS Google Scholar - Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J . Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem 2003; 278: 30772–30780.
Article Google Scholar - Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 2006; 103: 18154–18159.
Article CAS Google Scholar - Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 2007; 27: 912–925.
Article CAS Google Scholar - Ozer A, Wu LC, Bruick RK . The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF). Proc Natl Acad Sci USA 2005; 102: 7481–7486.
Article CAS Google Scholar - Takeda K, Fong GH . Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension 2007; 49: 178–184.
Article CAS Google Scholar - Bruick RK . Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev 2003; 17: 2614–2623.
Article CAS Google Scholar - Hagen T, Taylor CT, Lam F, Moncada S . Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 2003; 302: 1975–1978.
Article CAS Google Scholar - Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK . FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 2002; 16: 1466–1471.
Article CAS Google Scholar - Mahon PC, Hirota K, Semenza GL . FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001; 15: 2675–2686.
Article CAS Google Scholar - Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B . Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell 2003; 14: 3470–3481.
Article CAS Google Scholar - Sandau KB, Zhou J, Kietzmann T, Brune B . Regulation of the hypoxia-inducible factor 1alpha by the inflammatory mediators nitric oxide and tumor necrosis factor-alpha in contrast to desferroxamine and phenylarsine oxide. J Biol Chem 2001; 276: 39805–39811.
Article CAS Google Scholar - Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004; 118: 781–794.
Article CAS Google Scholar - Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275: 25130–25138.
Article CAS Google Scholar - Schroedl C, McClintock DS, Budinger GR, Chandel NS . Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Heart Circ Physiol 2002; 283: L922–L931.
CAS Google Scholar - Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ . Regulation of hypoxia-inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood 2001; 98: 296–302.
Article CAS Google Scholar - Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS . Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem 2004; 279: 40337–40344.
Article CAS Google Scholar - Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005; 7: 77–85.
Article CAS Google Scholar - Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem 2007; 282: 3293–3301.
Article CAS Google Scholar - Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J . Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem 2007; 282: 4524–4532.
Article CAS Google Scholar - Lu H, Forbes RA, Verma A . Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 2002; 277: 23111–23115.
Article CAS Google Scholar - Huang LE, Gu J, Schau M, Bunn HF . Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin–proteasome pathway. Proc Natl Acad Sci USA 1998; 95: 7987–7992.
Article CAS Google Scholar - Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ . Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J 2001; 20: 5197–5206.
Article CAS Google Scholar - Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 2004; 279: 38458–38465.
Article CAS Google Scholar - Huang J, Zhao Q, Mooney SM, Lee FS . Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem 2002; 277: 39792–39800.
Article CAS Google Scholar - Li D, Hirsila M, Koivunen P, Brenner MC, Xu L, Yang C et al. Many amino acid substitutions in a hypoxia-inducible transcription factor (HIF)-1alpha-like peptide cause only minor changes in its hydroxylation by the HIF prolyl 4-hydroxylases: substitution of 3,4-dehydroproline or azetidine-2-carboxylic acid for the proline leads to a high rate of uncoupled 2-oxoglutarate decarboxylation. J Biol Chem 2004; 279: 55051–55059.
Article CAS Google Scholar - Pereira T, Zheng X, Ruas JL, Tanimoto K, Poellinger L . Identification of residues critical for regulation of protein stability and the transactivation function of the hypoxia-inducible factor-1alpha by the von Hippel–Lindau tumor suppressor gene product. J Biol Chem 2003; 278: 6816–6823.
Article CAS Google Scholar - Chan DA, Sutphin PD, Yen SE, Giaccia AJ . Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol 2005; 25: 6415–6426.
Article CAS Google Scholar - Takeda K, Cowan A, Fong GH . Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 2007; 116: 774–781.
Article CAS Google Scholar - Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008 (in press).
- Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell 2005; 17: 503–512.
Article CAS Google Scholar - Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J . HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 2003; 22: 4082–4090.
Article CAS Google Scholar - Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W et al. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol 2006; 41: 68–77.
Article CAS Google Scholar - Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M et al. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci 2003; 116: 1319–1326.
Article CAS Google Scholar - Soilleux EJ, Turley H, Tian YM, Pugh CW, Gatter KC, Harris AL . Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 2005; 47: 602–610.
Article CAS Google Scholar - Metzen E, Stiehl DP, Doege K, Marxsen JH, Hellwig-Burgel T, Jelkmann W . Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: identification of a functional hypoxia-responsive element. Biochem J 2005; 387: 711–717.
Article CAS Google Scholar - Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO et al. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem J 2005; 390: 189–197.
Article CAS Google Scholar - Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T et al. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J 2004; 381: 761–767.
Article CAS Google Scholar - D'Angelo G, Duplan E, Boyer N, Vigne P, Frelin C . Hypoxia up-regulates prolyl hydroxylase activity: a feedback mechanism that limits HIF-1 responses during reoxygenation. J Biol Chem 2003; 278: 38183–38187.
Article CAS Google Scholar - Stiehl DP, Wirthner R, Koditz J, Spielmann P, Camenisch G, Wenger RH . Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. J Biol Chem 2006; 281: 23482–23491.
Article CAS Google Scholar - Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J . Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem 2007; 282: 1788–1796.
Article CAS Google Scholar - To KK, Huang LE . Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol Chem 2005; 280: 38102–38107.
Article CAS Google Scholar - McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM . Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 2006; 281: 24171–24181.
Article CAS Google Scholar - Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 2004; 117: 941–952.
Article CAS Google Scholar - Barth S, Nesper J, Hasgall PA, Wirthner R, Nytko KJ, Edlich F et al. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol 2007; 27: 3758–3768.
Article CAS Google Scholar - Hopfer U, Hopfer H, Jablonski K, Stahl RA, Wolf G . The novel WD-repeat protein Morg1 acts as a molecular scaffold for hypoxia-inducible factor prolyl hydroxylase 3 (PHD3). J Biol Chem 2006; 281: 8645–8655.
Article CAS Google Scholar - Arquier N, Vigne P, Duplan E, Hsu T, Therond PP, Frelin C et al. Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem J 2006; 393: 471–480.
Article CAS Google Scholar - Frei C, Galloni M, Hafen E, Edgar BA . The Drosophila mitochondrial ribosomal protein mRpL12 is required for Cyclin D/Cdk4-driven growth. EMBO J 2005; 24: 623–634.
Article CAS Google Scholar - Frei C, Edgar BA . Drosophila cyclin D/Cdk4 requires Hif-1 prolyl hydroxylase to drive cell growth. Dev Cell 2004; 6: 241–251.
Article CAS Google Scholar - Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA 2006; 103: 654–659.
Article CAS Google Scholar - Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS . A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 2007; 110: 2193–2196.
Article CAS Google Scholar - Erez N, Milyavsky M, Eilam R, Shats I, Goldfinger N, Rotter V . Expression of prolyl-hydroxylase-1 (PHD1/EGLN2) suppresses hypoxia inducible factor-1alpha activation and inhibits tumor growth. Cancer Res 2003; 63: 8777–8783.
CAS PubMed Google Scholar - Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N . Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int J Cancer 2006; 118: 1144–1153.
Article CAS Google Scholar - Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 2005; 8: 155–167.
Article Google Scholar - Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler III AA . Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res 2006; 98: 133–140.
Article CAS Google Scholar - Terkhorn SP, Bohensky J, Shapiro IM, Koyama E, Srinivas V . Expression of HIF prolyl hydroxylase isozymes in growth plate chondrocytes: relationship between maturation and apoptotic sensitivity. J Cell Physiol 2007; 210: 257–265.
Article CAS Google Scholar - Fu J, Menzies K, Freeman RS, Taubman MB . EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J Biol Chem 2007; 282: 12410–12418.
Article CAS Google Scholar - Irwin R, LaPres JJ, Kinser S, McCabe LR . Prolyl-hydroxylase inhibition and HIF activation in osteoblasts promotes an adipocytic phenotype. J Cell Biochem 2007; 100: 762–772.
Article CAS Google Scholar - Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH . Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol 2006; 26: 8336–8346.
Article CAS Google Scholar - Warnecke C, Griethe W, Weidemann A, Jurgensen JS, Willam C, Bachmann S et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J 2003; 17: 1186–1188.
Article CAS Google Scholar - Willam C, Masson N, Tian YM, Mahmood SA, Wilson MI, Bicknell R et al. Peptide blockade of HIFalpha degradation modulates cellular metabolism and angiogenesis. Proc Natl Acad Sci USA 2002; 99: 10423–10428.
Article CAS Google Scholar - Nangaku M, Izuhara Y, Takizawa S, Yamashita T, Fujii-Kuriyama Y, Ohneda O et al. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol 2007; 27: 2548–2554.
Article CAS Google Scholar - Hsieh MM, Linde NS, Wynter A, Metzger M, Wong C, Langsetmo I et al. HIF prolyl hydroxylase inhibition results in endogenous erythropoietin induction, erythrocytosis, and modest fetal hemoglobin expression in rhesus macaques. Blood 2007; 110: 2140–2147.
Article CAS Google Scholar
Acknowledgements
Publication cost of this article is supported by grants-in-aid to G-HF from the American Heart Association and March of Dimes.
Author information
Authors and Affiliations
- Department of Cell Biology, Center for Vascular Biology, University of Connecticut Health Center, Farmington, CT, USA
G-H Fong & K Takeda
Corresponding author
Correspondence toG-H Fong.
Additional information
Edited by N Chandel
Rights and permissions
About this article
Cite this article
Fong, GH., Takeda, K. Role and regulation of prolyl hydroxylase domain proteins.Cell Death Differ 15, 635–641 (2008). https://doi.org/10.1038/cdd.2008.10
- Received: 18 October 2007
- Revised: 07 January 2008
- Accepted: 08 January 2008
- Published: 15 February 2008
- Issue Date: April 2008
- DOI: https://doi.org/10.1038/cdd.2008.10