Alterations of brain and cerebellar proteomes linked to Aβ and tau pathology in a female triple-transgenic murine model of Alzheimer's disease (original) (raw)
Main
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that leads to cognitive and behavioral impairments. The pathological hallmarks of AD are neuritic plaques mainly composed of the β_-amyloid (A_β) peptide as well as intraneuronal neurofibrillary tangles (NFT) made of aggregated hyperphosphorylated microtubule-associated protein (MAP) tau (h-tau). Extracellular amyloid plaques are formed by the aggregation of the A_β_1–40 and A_β_1–42 peptides, fragments obtained from the proteolytic cleavage of the full-length amyloid precursor protein (APP) through the activity of the _β_- and γ_-secretase enzymes. This process, recapitulated in the ‘amyloid cascade hypothesis’, is thought to be the main mechanism leading to the synaptic loss and cognitive impairment observed in AD patients.1 A_β peptides also accumulate intraneuronally where they promote mitochondrial and proteasome dysfunction as well as calcium dyshomeostasis, and also facilitate the hyperphosphorylation of MAP tau thereby promoting NFT formation (reviewed in LaFerla et al.2). Only 1% of AD cases are familial, whereas most AD cases are sporadic. Familial AD (FAD) is linked to the presence of autosomal dominant mutations in at least three genes encoding the amyloid precursor protein (APP) and the presenilins 1 and 2 (PS1 and PS2); two components of the _γ_-secretase complex that are located on chromosomes 21, 14, and 1.3 These mutations lead to an increase in A_β_1–42 production. By contrast, the link between tau protein mutations and NFT formation in the AD brain is still unknown; however, mutant P301L tau leads to its hyperphosphorylation and NFT production in frontotemporal dementia as well as in a form of parkinsonism linked to a mutation found in chromosome 17.4 Thus, although not specific for AD, the P301L tau mutation is key to model tau neuropathology in transgenic animal models.
Recently, a triple-transgenic mouse model of AD (3 × Tg-AD; expressing mutant PS1M146V, APPswe, and tauP301L) has been developed.5 This mouse exhibits both A_β_ and h-tau alterations, thereby recapitulating most of the pathological features of the AD brain.
In recent years, several studies have described the differential protein expression profile in the brain of transgenic AD models (reviewed in Sowell et al.6) and a recent study has analyzed protein alterations occurring in male 3 × Tg-AD mice.7 In contrast, the cerebellar proteome in samples from AD mice or AD patients has been less investigated, most likely because the prevailing assumption is that this central nervous system (CNS) area is relatively unaffected by AD. However, it must be noted that some studies indicate that also the AD cerebellum shows some signs of morphological and metabolic dysfunctions.8
In this study, we performed a proteomic analysis in brains and cerebella of 14-month-old 3 × Tg-AD and control (CTRL) presenilin-1 knock-in (PS1-KI) female mice using two-dimensional gel electrophoresis (2D-E) and protein identification by mass spectrometry (MS). PS1-KI mice harboring the FAD-related M146V PS1 mutation do not express mutant APP or h-tau, do not show any AD-related cognitive decline nor A_β_- or tau-mediated pathology,9 thereby providing an adequate control to investigate the specific impact of the AD-like A_β_ and tau pathology occurring in 3 × Tg-AD mice. Finally, using the Protein Analysis Through Evolutionary Relationship (PANTHER) application system and Ingenuity Pathway Analysis (IPA), we inferred the functional role and networks linked to the proteins that we found differentially expressed.
Results
Protein expression profile and identification of differentially expressed proteins in the brain and cerebellum of PS1-KI and 3 × Tg-AD mice
2D-E-based analysis of protein expression profile of brain and cerebellum in PS1-KI (CTRL) and 3 × Tg-AD mice showed that 2D-E gel protein-spot patterns were qualitatively and quantitatively similar across all the analyzed gels. On average, we found 300 and 200 spots per gel in the brain and cerebellum, respectively. Of these genes, 86% (for the PS1-KI mice) and 96% (for 3 × TG-AD mice) were present across the four maps obtained from these two CNS regions. A subsequent analysis was performed only on common spots. Statistical analysis of the densitometry data allowed the identification of differentially expressed protein spots among PS1-KI and 3 × Tg-AD maps. As shown in Figure 1, we found 35 (in the whole brain) and 24 (in the cerebellum) differentially expressed spots that reached statistical significance (P<0.05).
Figure 1
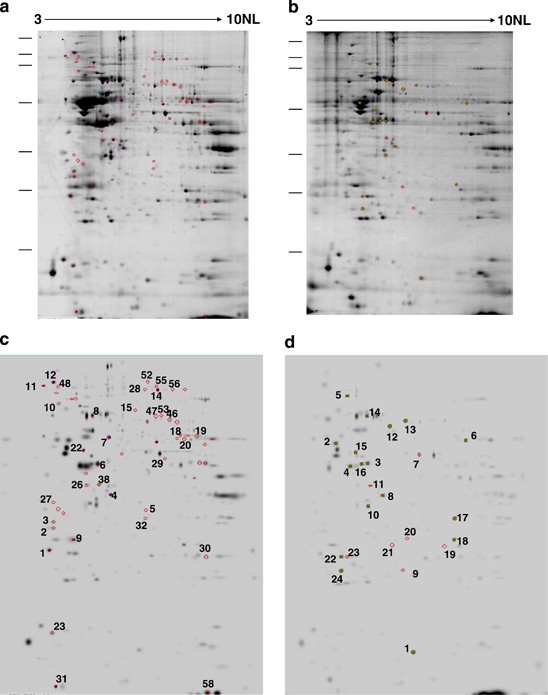
Representative 2D-E image obtained from either brains (a) or cerebella (b) of CTRL mice. The picture depicts the 2D-E gels obtained from CTRL samples using a nonlinear pH range of 3–10 in the first dimension (11 cm strips) and SDS-PAGE (12%) in the second. Differentially expressed proteins in brains (c) and cerebella (d) of 3 × Tg-AD versus CTRL mice. The picture depicts spots with a density that was significantly different (P<0.05) when comparing CTRL versus 3 × Tg-AD mice. Excised spots are numbered
We then identified the differentially expressed proteins. Peptide mass fingerprinting (PMF) by matrix-assisted laser desorption ionization-time of flight MS (MALDI-TOF-MS) analysis resulted in the identification of 26 (for the whole brain) and 14 (for the cerebellum) proteins (Tables 1 and 2). For protein identification, we considered only the proteins that can be resolved by the used electrophoretic procedures. In proteomic studies, efforts have been made to standardize results of PMF experiments. To that aim, we calculated the hit ratio (HR), the excess of limit-digested peptides (ELDP), the mass coverage (MC), and the peptide mass fingerprint score (PMFscore) as quality metrics for each mass spectrum that we have submitted for search in the database. To evaluate the quality of protein identification by PMF, we applied the methodological approach proposed by Stead et al.10 Using a large protein data set, Stead et al. showed that a specificity of 99% and a sensitivity of 78% in protein identification are accomplished when the PMFscore is higher than 79 and such cutoff was implemented in our study. Only in the case of four brain (spots 2, 9, 23, and 30) and four cerebellar (spots 19, 22, 23, and 24) proteins we calculated a PMFscore lower than 79. MS/MS experiments were then performed to validate the PMF data. Sequences of the identified peptides, the corresponding Mascot identification scores, as well as the peptide scores of PSD-MALDI-TOF-MS analysis of at least two of the most abundant signals detected in each protein spectrum are shown in Tables 1 and 2. In all cases, MS/MS analysis of the selected peptides confirmed the identifications obtained with PMF experiments.
Table 1 Differentially expressed proteins in the brain of PS1KI (CTRL) and 3 × Tg-AD mice
Table 2 Differentially expressed proteins in the cerebellum of PS1KI (CTRL) and 3 × Tg-AD mice
Theoretical and experimental molecular weights (MWs) as well as isoelectric points (pIs) calculated from 2D-E maps are shown in Tables 1 and 2. The values of MW and pI of each protein corresponded roughly to its position on the 2D-E gel. Deviations from theoretical MW and pI were found to be lower than 30% in several cases. In the case of brain spot 11, identified as Drebrin 1, we observed a significant deviation from its theoretical MW; this is however compatible with its apparent MW of ∼130 kDa in sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) as previously reported.11 Some representative differentially expressed proteins are shown in Figure 2a (for whole brain) and b (for the cerebellum). The mean density ratios (_n_=4) calculated for each identified proteins are depicted in Tables 1 and 2. Out of the 26 identified brain proteins, 23 were significantly downregulated (Table 1).
Figure 2
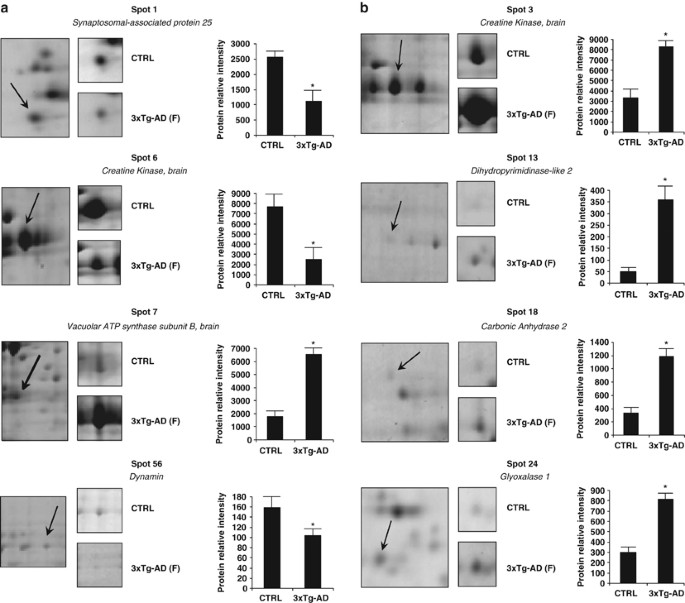
Differential protein expression in brains (a) and cerebella (b) of CTRL and 3 × TG-AD mice. The picture depicts representative subsections of 2D-E images. In each panel, bar charts show relative intensities (RI) (means±s.d.; _n_=4) of protein spots obtained from CTRL and 3 × Tg-AD mice. *P<0.05
In contrast, all the identified cerebellar proteins were upregulated (Table 2). Interestingly, some of the cerebellar upregulated proteins, Rho-GDP dissociation inhibitor 1 (ARHGDA), brain creatine kinase (CKB), dihydropyrimidinase-like2 protein (DPYSL2), enolase 1 (α) (ENO1), glutamate dehydrogenase 1 (GLUD1), isocitrate dehydrogenase 3 (NAD+) α (IDH3A), and lactate dehydrogenase B (LDHB) were, on the contrary, significantly downregulated in the whole brain.
Functional and protein network analysis
To investigate the functional significance of the protein differentially expressed, we performed a functional classification by using PANTHER application system (Figure 3). In the whole brain (Figure 3a), this analysis resulted in four clusters related to carbohydrate metabolism, cell structure and motility, transport, and intracellular protein traffic, whereas in the cerebellum (Figure 3b) we found two clusters related to carbohydrate metabolism and protein metabolism and modification. Further analysis in the Swissprot database (www.expasy.org) showed that six of the downregulated brain proteins, mitochondrial aconitase 2 (ACO2), GLUD1, mitochondrial glycerol-3-phosphate dehydrogenase 2 (GPD2), IDH3A, oxoglutarate dehydrogenase-like protein (OGDHL), ubiquinol-cytochrome c reductase core protein I (UQCRC1) are localized in mitochondria in which they function as enzymes involved in the tri-carboxylic acid (TCA) cycle and in oxidative phosphorylation.
Figure 3
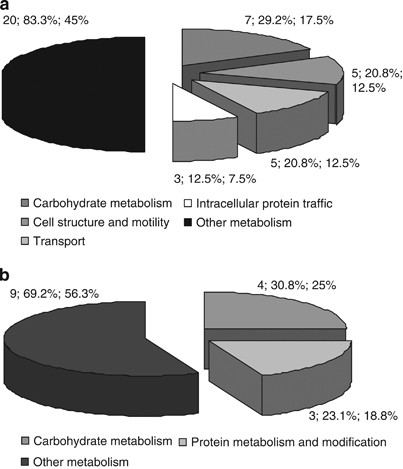
Functional classification of identified proteins in brains (a) and cerebella (b) of 3 × Tg-AD mice. Pie charts show PANTHER classifications made according to the biological functions associated to each protein. Numbers indicate the number of proteins belonging to the specified functional category, the percentage of protein hit against the total number of proteins, as well as the percentage of protein hit against the total number of processed hits
To obtain further insight of potential biological mechanisms involving the differentially expressed proteins, we performed an Ingenuity Knowledge Base search using the IPA application. This tool builds protein networks based on known direct and indirect interactions described in the literature and defines common functional and canonical pathways. The IPA-generated networks show the identified proteins along with other interacting proteins and/or endogenous chemicals.
A search in the Ingenuity Knowledge Base identified two ‘high score’ overlapping networks (Figures 4a and b; Supplementary Table S1). These networks contain the following 16 identified proteins (focus genes): ACO2; ARHGDIA; CKB; DBN1; dynamin 1 (DNM1); DPYSL2; GLUD1; GPD2; heat shock protein 90 kDa β (Grp94) member 1; LDHB; neurofilament, medium polypeptide (NEFM); phosphatidylinositol transfer protein-α (PITPNA); synaptosomal-associated protein, 25 kDa (SNAP25); synuclein-α (SNCA); triosephosphate isomerase 1 (TPI1); and tropomyosin 1 (TPM1); and seven, ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A (ATP6V1A); ATPase, H+ transporting, lysosomal 56/58 kDa, V1 subunit B2; ENO1; hemoglobin-β; IDH3A; UQCRC1; and WD repeat domain 1 (WDR1). Two molecules, dehydroisoandrosterone and peroxisome proliferator-activated receptor γ-co-activator 1_α_, were found common between these networks.
Figure 4
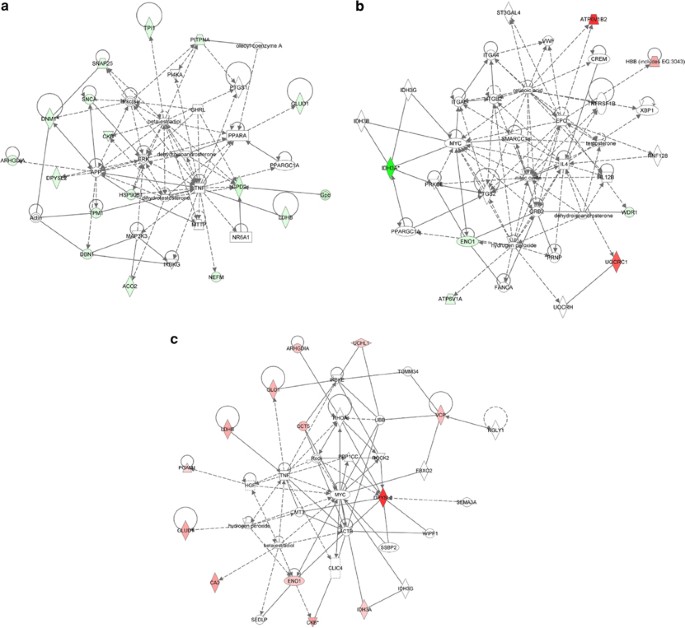
Ingenuity Pathway Analysis of differentially expressed proteins: network interaction for brain (a and b) and cerebellar (c) proteins. Diagrams show focus genes (the identified proteins found differentially expressed) that are depicted in green (downregulated) or red (upregulated). Solid and dashed connecting lines indicate the presence of direct and indirect interactions, respectively. Modulatory roles of proteins and/or endogenous chemicals on the expression of other proteins are indicated by arrows
The main biological functions associated with the first network (score=41) are related to lipid metabolism and molecular transport. However, IPA classified six mapped proteins, DNM1, SNCA, DPYSL2, NEFM, DBN1, and SNAP25, as specifically involved in the development and functional maintenance of the nervous system as well as in neurodegeneration. The second network (score=15) is significantly associated with tissue morphology and cell damage or degeneration in the form of cell atrophy and swelling. Overall, the top canonical pathways associated with the altered protein expression found in whole 3 × Tg-AD brains were the ones related to glycolysis/gluconeogenesis, mitochondrial metabolism, and oxidative phosphorylation.
In the case of 3 × Tg-AD cerebella, we found one network (Figure 4c; Supplementary Table S1; score=38) containing 13 focus proteins: ARHGDIA; carbonic anhydrase II (CA2); CKB; chaperonin-containing TCP1, subunit 5 (epsilon) (CCT5); DPYSL2; ENO1; glyoxalase I; GLUD1; IDH3A; LDHB; phosphoglycerate mutase 1 (brain) (PGAM1); ubiquitin carboxyl-terminal esterase L1 (UCHL1); and valosin-containing protein (VCP). This network is functionally associated to cancer, cellular growth and proliferation, as well as to cellular compromise.
Discussion
In our study, using a classical proteomic approach based on 2-DE and MS, we analyzed the differential protein expression associated with the combination of mutant PS1M146V, APPSwe, and tauP301L in the brain and cerebellum of 14-month-old 3 × Tg-AD female mice. To date, there is only one study describing the protein expression in the brain of the same model.7 In this study, Martin et al. evaluated the protein expression profile in the cortex and hippocampus, but not in the cerebellum, of 16-month-old male 3 × Tg-AD mice (for a detailed analysis of the differences between the two studies see Supplementary Figure S1 and Supplementary Table S2). In their study, Martin et al. proposed a gel-free proteomics platform that, compared to what we were able to identify, allows the identification of a higher number of differentially expressed proteins. However, it must be noted that although gel-free proteomics has a higher throughput and overcomes some limitations of gel-based proteomics (such as the poor separation of highly hydrophobic proteins or proteins showing extreme, low or high, pI values, and MW), 2D-E is currently considered the standard technique for studying the relative quantification of protein variants. In that respect, our findings are not merely reproducing what already described by Martin et al., but are novel and complementary to such data set. Furthermore, Martin et al. chose to analyze selected brain subregions such as the hippocampus and the cortex to provide insightful information on the regional progression of the AD-like pathology in 3 × Tg-AD mice. However, it should be noted that the 3 × Tg-AD mice do not show a clear reproducible specific regional pattern of amyloid deposition and tau pathology.12 Thus, to avoid a great deal of variability, we have chosen to analyze proteomic change in the whole brain.
Finally, it must be also emphasized that gender has a significant impact on the cognitive and neuropathological phenotype of AD model13 with female 3 × Tg-AD mice exhibiting a more aggressive disease progression.12 We therefore decided to investigate the whole brain and cerebellum of female 3 × Tg-AD mice at 14 months of age (m.o.a.) as 3 × Tg-AD mice show intraneuronal A_β_ accumulation at 3–4 m.o.a., amyloid plaques at 12 m.o.a. whereas they do develop NFTs at 12–15 m.o.a.14 We would also like to emphasize that, to date, this is the first study describing cerebellar proteomic changes of 3 × Tg-AD mice. Cerebellum is less affected than other CNS areas from AD and our data can therefore provide critical information on region-specific mechanisms that are counteracting the development of AD-related injury. Finally, we chose to investigate age-matched female PS1-KI mice as CTRL model because mutant PS1 mice show elevated levels of murine A_β_ as well as dyshomeostasis of intracellular Ca2+,15 but do not exhibit any A_β_- or tau-dependent neuronal degeneration or cognitive impairment.9 In fact, given the amino-acid composition of murine amyloid as well as the different susceptibility of murine APP to the enzymatic cleavage by BACE1, the peptide is unable to aggregate to form plaques or intracellular deposits. Therefore, the mere expression of mutant PS1 has no effects on parenchymal or intraneuronal amyloid deposition,16, 17 and it is only when human mutant PS1 is co-expressed along with human mutant APP that amyloid-driven overload and pathology can be observed and studied in murine AD models.17
Overall, our data are often in agreement with the expression levels of several proteins found in brains of AD patients and/or AD animal models.6, 18
To further elucidate the functional implications of the changes we detected, we plot the data into canonical pathways as suggested by the PANTHER application system. This analysis indicates that the proteins we found to be differentially expressed in the whole brain are functionally organized in at least four clusters, namely carbohydrate metabolism, cell structure and motility, transport, and intracellular protein traffic (Figure 4a).
Impairment of brain metabolism has been recognized as a hallmark of AD and the reduction of glucose utilization is paralleled by a decrease in the expression of glycolytic enzymes.19
In our study, we observed a significant downregulation of proteins such as TPI1 and ENO1, which could explain an impaired glucose metabolism, similarly to what reported in AD patients. Our analysis also reveals the downregulation of mitochondrial proteins like FAD-dependent GDP2, ACO2, two isoforms of the α subunit of NAD+-dependent isocitrate dehydrogenase complex (IDH3A), as well as OGDHL, a brain-specific isoenzyme of 2-oxoglutarate dehydrogenase (OGDH). ACO2, IDH3, and OGDH are also key enzymes in the TCA cycle. IDH3 activity regulates OGDH that is the primary site of control of the metabolic flux through the TCA cycle. Decreased IDH and OGDH activities and expression levels have been observed in AD brains.20 It is interestingly to note that the downregulation of both OGDH and mitochondrial GLUD1, key enzymes participating in glutamate (Glu) metabolism, can result in deranged Glu production of and the impairment of glutamatergic neurotransmission. Furthermore, CKB was significantly downregulated in agreement with what reported in AD brains.21 This downregulation might result in decreased extra-mitochondrial production of ATP and its altered distribution to specific intracellular compartments.
Taken together, these findings suggest that A_β_ pathology and tau pathology promote a significant impairment of energy metabolism in brains of female 3 × Tg-AD mice. Our results pinpoint the TCA cycle and mitochondrial dysfunctions as key targets of the AD-like pathology of these mice. Gender might have a role here as several of these proteins were not found differentially expressed in 16-month-old male 3 × Tg-AD mice (Supplementary Table 2).
Interestingly, several evidences indicate that mitochondrial dysfunction can greatly affect synapse functioning (reviewed in Reddy and Beal22).
A more direct link to altered synaptic activities is also suggested by what we found as far as the downregulation of proteins that have a key role in synaptic functioning and synapse formation (DBN1, DPYSL2, SNAP25) as well as in endocytosis and synaptic vesicle recycling (ATP6V1A, DYN1, PITPNA). DBN1 modulates synaptic plasticity and the shaping of the cytoskeleton by interacting with actin, thereby regulating the morphogenesis, patterning, as well as the maintenance of dendritic spines in neurons whereas DPYSL2 downregulation might be associated to neuronal and synaptic loss as the protein is involved in axon guidance and growth, neurite development, and neuronal polarization. Furthermore, decreased DYN1 and PITPNA expression leads to the impairment of synaptic release of neurotransmitters. It is interesting to note that the downregulation of DYN1, DBN1, DPYSL2, and SNAP25 has been reported in brains of AD patients.21, 23, 24, 25 Synaptic loss is the major neurobiological substrate of cognitive dysfunction in AD26 and, taken together, our results suggest a significant impairment of synaptic functions and plasticity in the late stage AD-like pathology observed in 3 × Tg-AD mice.
Loss of functional synapses in AD also appears to be related to amyloid- and tau-driven cytoskeleton alterations.27 Interestingly, our results indicate that several components of the neuronal cytoskeleton, such as TPM1, NEFM, and WDR1, are significantly downregulated in the 3 × Tg-AD mice. ARHGDIA is indirectly related to the rearrangement of the cytoskeleton. In fact, ARHGDIA inhibits Rho GTP-ase and its main effectors serine/threonine Rho kinases that promote the development of the growth cone and rearrange the cytoskeleton.28 The decreased expression of ARHGDIA might be related to an increase of Rho GTP-ase and Rho-kinase activities followed by an inhibition of neurite outgrowth. Cytoskeletal alterations found in AD patients have been also linked to deranged chaperone functions and protein misfolding. The chaperon protein Hsp90 has a pivotal role in aberrant tau degradation and/or tau refolding.29 It is still unclear whether tau pathology results from toxic effects of abnormally phosphorylated MAP tau or loss of its physiological function in AD. Possible explanations could arise by understanding the relationship between Hsp90 and MAP tau processing.
Interestingly, in silico network analysis further substantiates the idea that APP and A_β_ modulate the expression of several identified synaptic and cytoskeletal proteins (Figure 4a), suggesting a key role of APP aberrant processing in the observed proteomics alterations. For example, A_β_ promotes a significant downregulation of DBN130 and DYN131 in cell cultures whereas intracellular levels of APP intracellular domain have been associated to deranged function of DPYSL2.32 In summary, all these changes offer novel mechanisms to further understand the synaptic toxicity of A_β_.
In contrast to what is observed in the brain, all the proteins found differentially expressed in the cerebellum resulted to be upregulated whereas seven of these were actually downregulated in whole brain samples (i.e., ARHGDA, CKB, DPYSL2, ENO1, GLUD1, IDH3A, and LDHB). These results suggest that mutant APP and/or tau affect similar pathways in brain and cerebellum but in opposite direction. Only few data are currently available on proteome of the cerebellum of AD patients or AD mouse models. The cerebellum is thought to be less affected than the cerebral cortex and hippocampus by AD; however, pathological cerebellar changes are found in advanced stages of AD.8
Functional analysis of overexpressed cerebellar proteins in 3 × Tg-AD mice indicates two main protein clusters related to protein processing and carbohydrate metabolism (Figure 3b).
Among the proteins associated with protein metabolism, we found VCP, UCHL1, and mKIAA0098 protein, also known as CCT5. All these proteins are involved in the chaperone–proteasome system and have been linked to AD neurodegeneration.33 Increased VCP expression levels or activity may enhance the protein degradation mediated by the ubiquitin–proteasome system (UPS) whereas the main function of UCHL1, an important component of the UPS, pertains the recycling of monoubiquitin and its stabilization. Interestingly, UCHL1 has been found to be decreased and its level is inversely proportional to the number of NFTs in AD brain.34 UCHL1 is also required for normal synaptic function and rescues A_β_-related synaptic dysfunction in transgenic AD mice.35 CCT has been found significantly reduced in AD brains36 and may be involved in AD-related cytoskeletal abnormalities, thus it is intriguing to hypothesize that the cerebellar upregulation we find in 3 × Tg-AD may serve a role in counteracting these cytoskeletal alterations. This hypothesis is further supported by the increased expression levels of ARGDHA and DPYSL2. As discussed above, these proteins have a key role in the maintenance of the neuronal cytoskeleton and promotion of neuronal development. Interestingly, ARHGDIA also possesses antiapoptotic activity37 and, thus, increased ARHGDIA levels might exert an important neuroprotective effect on the cerebellum.
GLYO1 and CA2 were also found specifically upregulated in the cerebellum. GLYO1 is one of the components of the glyoxalase system that detoxifies _α_-ketoaldehydes, preventing the formation of advanced glycation end products that are known to generate oxidative stress and reduce proteolysis of carbonylated MAP-tau. CA2 is a CA isozyme and decreased brain CA levels have been associated with the alteration of synaptic spines and dendrite loss observed during aging and AD.38
It is again intriguing to speculate that the cerebellar overexpression of CKB, IDH3A, LDHB, and PGAM1 as well as mitochondrial proteins such as IDH3A and GLUD1 might compensate for an AD-related deficit of energy metabolism that could otherwise develop in this region.
Overall, our results suggest that the cerebellum largely differs from brain in response to the pro-AD environment present in the 3 × Tg-AD mice. Network analysis by IPA (Figure 4c) shows networks associated with cellular growth and proliferation. Key nodes of such networks are the MYC oncogene and the hepatocyte growth factor, a protein that stimulates cell proliferation. The identified network indicates an important inhibitory effect of ARHGDIA on RHOA and ROCK2 and also includes actin and ubiquitin, suggesting that, in response to pro-AD factors, the cerebellum sets in motion a reorganization of the cytoskeleton and enhances clearance of misfolded proteins by activating the UPS.
In conclusion, our proteomic study in the brain and cerebellum of 3 × Tg-AD mice show an interesting divergence of effects between these two CNS regions. In the brain, we observe a significant downregulation of synaptic, cytoskeletal, and mitochondrial proteins suggesting that synaptic and mitochondrial dysfunction are having a key role in the late stage of the AD-like pathology of this region. In contrast, in the cerebellum, we find an upregulation of proteins that are involved in energy metabolism, clearance of misfolded protein, and detoxification. These findings are particularly intriguing as they can shed new light on endogenous mechanisms promoted by the cerebellum to counteract the pathogenic actions of A_β_ and _P_-tau and ultimately offer novel targets for therapeutic intervention.
Materials and Methods
Animals
Female, 14-month-old PS1-KI (CTRL, _n_=4) and 3 × Tg-AD (_n_=4) mice were kept in a temperature-controlled room at 25°C under a 12-h light/dark cycle and fed ad libitum with a commercial pelleted feed and water.
At the age of 14 months, mice were anesthetized and killed by decapitation. Whole brains and cerebella were excised, weighed, immediately frozen in liquid nitrogen, and then stored at −80°C for later processing.
All experiments were performed in accordance with general guidelines regarding animal experiments and approved by our institutional committee for animal care.
Protein extraction, sample preparation, and two-dimensional gel electrophoresis
2D-E was performed with little modification of previous protocols.39 In brief, frozen brain/cerebellum tissue (at least 40 mg) was homogenized in 1 ml of 8.3 M urea, 2 M thiourea, 2% 3-((3-cholamidopropyl)dimethylammonio)-1-propanesulfonate hydrate, 1% dithiothreitol (DTT; Sigma-Aldrich, Schnelldorf, Germany), 2% IPG buffer (pH 3–10), using a polytron. Crude extracts were vigorously shaken for 30 min at 4°C, followed by a 30 min centrifugation at 10 000 × g. Determination of protein concentration was performed using the Bio-Rad DC method (Bio-Rad Laboratories, Hercules, CA, USA). Total protein extracts were prepared for each animal and each individual sample was assessed separately. Samples of 250 _μ_g protein were applied to immobilized (pH 3–10) nonlinear gradient strips (11 cm; Bio-Rad Laboratories). For each sample, triplicate runs were performed as independent experiments. Samples of 1 mg protein were used for preparative gels (immobilized pH 3–10 nonlinear gradient strips, 17 cm). Focusing started at 200 V, with the voltage being gradually increased to 3500 V and then kept constant for a further 66 500 V (PROTEAN IEF System; Bio-Rad Laboratories). Before SDS-PAGE, the IPG strips were incubated for 15 min with a solution of Tris-Cl buffer (pH 8.8), urea (6 M), glycerol (30%, v/v; Sigma-Aldrich), SDS (2%, w/v; Sigma-Aldrich), and DTT (1%, w/v). Strips were then equilibrated for further 15 min in the same buffer containing iodoacetamide (IAA, 5%, w/v; Sigma-Aldrich) instead of DTT. The second-dimensional separation was performed in 12% SDS-polyacrylamide gels. After protein fixation (in 40% methanol, 7% acetic acid aqueous solution), gels were stained with colloidal Coomassie blue (Sigma-Aldrich).
Protein visualization and image analysis
Electronic images of gels were acquired by a calibrated densitometer (GS-800; Bio-Rad Laboratories) and analyzed using PDQuest software (Bio-Rad Laboratories). Molecular masses were determined by running standard protein markers, covering the range of 10–200 kDa. The pI values used were those given by the supplier of the IPG strips. Background was automatically removed by the software on the basis of selected faint spots manually identified on the master gel.
Statistical analysis
The relative intensity (RI) for each spot corresponding to normalized values was calculated accordingly with the following formula:  where _V_i is the volume of the individual spot, n the number of detected spots, and
where _V_i is the volume of the individual spot, n the number of detected spots, and  the sum of the volumes of all the matched spots. For each matchset analysis, maps corresponding to protein extracts from animals in the same experimental group were organized into ‘Replicate Groups’ and named as either (CTRL) or (3 × Tg). This allowed us to carry out an automatic statistical analysis of densitometric data, expressed as normalized spot densities by using the Student's _t_-test. Statistical significance for differentially expressed spots was assumed with _P_-values lower than 0.05. The mean density ratios of differentially expressed proteins were then calculated.
the sum of the volumes of all the matched spots. For each matchset analysis, maps corresponding to protein extracts from animals in the same experimental group were organized into ‘Replicate Groups’ and named as either (CTRL) or (3 × Tg). This allowed us to carry out an automatic statistical analysis of densitometric data, expressed as normalized spot densities by using the Student's _t_-test. Statistical significance for differentially expressed spots was assumed with _P_-values lower than 0.05. The mean density ratios of differentially expressed proteins were then calculated.
In-gel digestion and MALDI-TOF-MS analysis
Protein spots showing significant changes in mean volume were selected for spot picking, trypsin digestion, and MALDI-TOF-MS analysis. Selected protein spots were manually excised and stored at −80°C until digestion. Gel plugs were washed twice with water and 50% acetonitrile. The cysteine residues were reduced by adding 50 _μ_l DTT (20 mmol l−1; Sigma-Aldrich) at 57°C and alkylated by 50 _μ_l IAA (100 mmol l−1; Sigma-Aldrich) at room temperature. The gel slices washed three times for 10 min each with 200 _μ_l of 40 mmol l−1 ammonium bicarbonate in 50% acetonitrile, dehydrated with 100% acetonitrile, and then rehydrated with 20 _μ_l of a 10 mg l−1 trypsin (Sigma-Aldrich) solution in 50 mmol l−1 ammonium bicarbonate. Trypsin digestion was carried out at 37°C for 12 h. In-gel digested protein samples were concentrated and desalted by using ZipTip C18 (Millipore, Watford, Hertfordshire, UK) accordingly to the manufacturer's directions. Elution was performed with a saturated solution of _α_-cyano-4-hydroxycinnamic acid (50% acetonitrile/water; Sigma-Aldrich) directly on an MTP Ground Steel 384 target (Bruker Daltonics, Bremen, Germany). Mass spectra were acquired between 700 and 4000 Da on a MALDI-TOF mass spectrometer Reflex IV (Bruker Daltonics) operating in reflectron mode. A 50 pmol _μ_l−1 solution of angiotensin I (m/z 1296.68; Sigma-Aldrich), ACTH (fragment 18–39, m/z 2465.19; Sigma-Aldrich), bradikynin (fragment 1–9, m/z 1060.57; Sigma-Aldrich), and renin substrate (m/z 1758.93; Sigma-Aldrich) was used for the external calibration. Mass spectra were processed to generate peak lists using Flex Analysis version 2.0 (Bruker Daltonics). Automated peak detection was performed using the SNAP algorithm and a signal-to-noise threshold of 6. Internal calibration was based on trypsin autodigestion peptides. Database search was performed by the Mascot algorithm. The databases searched were NCBInr using the following input parameters: taxonomy=mus musculus; enzyme=trypsin; allowed missed cleavages=1; fixed modification=carbamidomethyl (Cys); variable modification=oxidation (Met); peptide tolerance=50 ppm; mass value=MH+, monoisotopic. For each spectrum, the HR, the ELDP, and the MC were calculated as previously reported10 as: HR=(_n_masses matched)/(_n_masses submitted); ELPD=(_n_matched peptides having no missed cleavages)−(_n_matched peptides containing a missed cleavage site); MC=(%sequence coverage/100) × MW(kDa). A PMFscore was calculated from these quality metrics as: PMFscore=(HR × 100)+MC+(ELPD × 10).
Post source decay (PSD) MALDI-TOF-MS/MS spectra of at least two most abundant ions present in PMF spectra of proteins with a PMFscore lower than 79 were acquired. MS/MS spectra were acquired by using the same MALDI-TOF mass spectrometer and the acquisition program FAST. Twelve fragment spectra were added to obtain each MS/MS spectrum and at least 300 shots were added for each fragment. The individual spectra were pasted together using the Bruker FAST software. Searches in the NCBInr or Swissprot databases were performed using the Mascot algorithm. Parent ion and fragment mass tolerances were of 50 p.p.m. and 1 Da, respectively.
Functional classification and protein network analysis
Functional classification of the identified proteins was performed using the PANTHER application system40 (http://www.pantherdb.org) that uses information on protein sequence to assign a gene to an ontology group on the basis of the Gene Ontology terms (http://www.geneontology.org).
In silico protein network analysis was instead performed using the IPA application (Ingenuity System, http://www.ingenuity.com). IPA constructs hypothetical protein interaction clusters on the basis of the Ingenuity Pathways Knowledge Base. A data set containing the protein identifiers of the proteins found changed and the corresponding expression value was uploaded into the application. The expression values were the mean density ratios calculated for each spots. IPA automatically converts the mean density ratios to fold changes. Direct and indirect relationships between the identified proteins were shown as networks on the base of all genes and endogenous chemicals present in the Ingenuity Knowledge Base. IPA computes a score for each identified network. These scores are derived from a _P_-value and indicate the likelihood that focus genes (i.e., the identified proteins within a specific network) are clustered together. Biological functions and canonical pathways overrepresented among the identified proteins were also assigned to networks stored in the Ingenuity Pathways Knowledge Base. Biological functions and canonical pathways were ranked in accordance to their significance within these networks. Significance was evaluated by exact Fisher's test. A _P_-value of 0.01 corresponding to a score of 2 was considered the cutoff for the analysis.
Abbreviations
3 × Tg-AD:
triple-transgenic mouse model of Alzheimer's disease
PS1-KI:
presenilin-1 knock-in mice
APP:
amyloid precursor protein
AD:
Alzheimer's disease
h-tau:
hyperphosphorylated tau
A_β_:
_β_-amyloid
NFT:
neurofibrillary tangle
MAP:
microtubule-associated protein
FAD:
familial AD
PS1 and PS2:
presenilins 1 and 2
CNS:
central nervous system
2D-E:
two-dimensional gel electrophoresis
MS:
mass spectrometry
PANTHER:
Protein Analysis Through Evolutionary Relationship
IPA:
Ingenuity Pathway Analysis
DTT:
dithiothreitol
SDS-PAGE:
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
IAA:
iodoacetamide
MALDI-TOF-MS:
matrix-assisted laser desorption ionization-time of flight MS
HR:
hit ratio
ELDP:
excess of limit-digested peptides
MC:
mass coverage
PMFscore:
peptide mass fingerprint score
PSD:
post source decay
ARHGDA:
rho-GDP dissociation inhibitor 1
CKB:
brain creatine kinase
DPYSL2:
dihydropyrimidinase-like2 protein
ENO1:
enolase 1 (α)
GLUD1:
glutamate dehydrogenase 1
IDH3A:
isocitrate dehydrogenase 3 (NAD+) α
LDHB:
lactate dehydrogenase B
ACO2:
mitochondrial aconitase 2
GPD2:
mitochondrial glycerol-3-phosphate dehydrogenase 2
OGDHL:
oxoglutarate dehydrogenase-like protein
UQCRC1:
ubiquinol-cytochrome c reductase core protein I
DNM1:
dynamin 1
∣NEFM:
neurofilament, medium polypeptide
PITPNA:
phosphatidylinositol transfer protein, α
SNAP25:
synaptosomal-associated protein, 25 kDa
SNCA:
synuclein-α
TPI1:
triosephosphate isomerase 1
TPM1:
tropomyosin 1
ATP6V1A:
ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A
WDR1:
WD repeat domain 1
CA2:
carbonic anhydrase II
CCT5, chaperonin-containing TCP1subunit 5 (epsilon)∣PGAM1:
phosphoglycerate mutase 1 (brain)
UCHL1:
ubiquitin C-terminal esterase L1
VCP:
valosin-containing protein
References
- Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Article CAS Google Scholar - LaFerla FM, Green KN, Oddo S . Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci 2007; 8: 499–509.
Article CAS Google Scholar - Hardy J . Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 1997; 20: 154–159.
Article CAS Google Scholar - Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702–705.
Article CAS Google Scholar - Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003; 39: 409–421.
Article CAS Google Scholar - Sowell RA, Owen JB, Butterfield DA . Proteomics in animal models of Alzheimer's and Parkinson's diseases. Ageing Res Rev 2009; 8: 1–17.
Article CAS Google Scholar - Martin B, Brenneman R, Becker KG, Gucek M, Cole RN, Maudsley S . iTRAQ analysis of complex proteome alterations in 3 × TgAD Alzheimer's mice: understanding the interface between physiology and disease. PLoS One 2008; 3: e2750.
Article Google Scholar - Larner AJ . The cerebellum in Alzheimer's disease. Dement Geriatr Cogn Disord 1997; 8: 203–209.
Article CAS Google Scholar - Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM . Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 2005; 45: 675–688.
Article CAS Google Scholar - Stead DA, Preece A, Brown AJ . Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol Cell Proteomics 2006; 5: 1205–1211.
Article CAS Google Scholar - Ishikawa R, Hayashi K, Shirao T, Xue Y, Takagi T, Sasaki Y et al. Drebrin, a development-associated brain protein from rat embryo, causes the dissociation of tropomyosin from actin filaments. J Biol Chem 1994; 269: 29928–29933.
CAS PubMed Google Scholar - Oh K-J, Perez SE, Lagalwar S, Vana L, Binder L, Muffson EJ . Staging of Alzheimer's pathology in triple transgenic mice: a light and electron microscopic analysis. Int J Alzheimer Dis 2010, 780102.
- Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC . Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol 2001; 158: 1173–1177.
Article CAS Google Scholar - Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM . Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 2003; 24: 1063–1070.
Article CAS Google Scholar - Smith IF, Green KN, LaFerla FM . Calcium dysregulation in Alzheimer's disease: recent advances gained from genetically modified animals. Cell Calcium 2005; 38: 427–437.
Article CAS Google Scholar - Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 1996; 383: 710–713.
Article CAS Google Scholar - Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997; 19: 939–945.
Article CAS Google Scholar - Butterfield DA, Boyd-Kimball D, Castegna A . Proteomics in Alzheimer's disease: insights into potential mechanisms of neurodegeneration. J Neurochem 2003; 86: 1313–1327.
Article CAS Google Scholar - Hoyer S, Nitsch R, Oesterreich K . Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J Neural Transm Park Dis Dement Sect 1991; 3: 1–14.
Article CAS Google Scholar - Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE . Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol 2005; 57: 695–703.
Article CAS Google Scholar - Schonberger SJ, Edgar PF, Kydd R, Faull RL, Cooper GJ . Proteomic analysis of the brain in Alzheimer's disease: molecular phenotype of a complex disease process. Proteomics 2001; 1: 1519–1528.
Article CAS Google Scholar - Reddy PH, Beal MF . Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med 2008; 14: 45–53.
Article CAS Google Scholar - Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis 2003; 12: 97–109.
Article CAS Google Scholar - Shim KS, Lubec G . Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci Lett 2002; 324: 209–212.
Article CAS Google Scholar - Lubec G, Nonaka M, Krapfenbauer K, Gratzer M, Cairns N, Fountoulakis M . Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down syndrome and Alzheimer's disease brain is downregulated at the mRNA and dysregulated at the protein level. J Neural Transm Suppl 1999; 57: 161–177.
CAS PubMed Google Scholar - Arendt T . Synaptic degeneration in Alzheimer's disease. Acta Neuropathol 2009; 118: 167–179.
Article Google Scholar - Bamburg JR, Bloom GS . Cytoskeletal pathologies of Alzheimer disease. Cell Motil Cytoskeleton 2009; 66: 635–649.
Article CAS Google Scholar - Govek EE, Newey SE, Van Aelst L . The role of the Rho GTPases in neuronal development. Genes Dev 2005; 19: 1–49.
Article CAS Google Scholar - Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J et al. The high-affinity HSP90–CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 2007; 117: 648–658.
Article CAS Google Scholar - Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 2007; 27: 796–807.
Article CAS Google Scholar - Kelly BL, Ferreira A . beta-Amyloid-induced dynamin 1 degradation is mediated by _N_-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem 2006; 281: 28079–28089.
Article CAS Google Scholar - Ryan KA, Pimplikar SW . Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J Cell Biol 2005; 171: 327–335.
Article CAS Google Scholar - Dai RM, Li CC . Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 2001; 3: 740–744.
Article CAS Google Scholar - Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS et al. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem 2004; 279: 13256–13264.
Article CAS Google Scholar - Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 2006; 126: 775–788.
Article CAS Google Scholar - Schuller E, Gulesserian T, Seidl R, Cairns N, Lube G . Brain t-complex polypeptide 1 (TCP-1) related to its natural substrate beta1 tubulin is decreased in Alzheimer's disease. Life Sci 2001; 69: 263–270.
Article CAS Google Scholar - Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF . Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol 2001; 3: 339–345.
Article CAS Google Scholar - Meier-Ruge W, Iwangoff PK . Neurochemical enzyme changes in Alzheimer's and Pick's disease. Arch Gerontol Geriatr 1984; 3: 161–165.
Article CAS Google Scholar - Fountoulakis M, Tsangaris GT, Maris A, Lubec G . The rat brain hippocampus proteome. J Chromatogr B Analyt Technol Biomed Life Sci 2005; 819: 115–129.
Article CAS Google Scholar - Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 2005; 33: D284–D288.
Article CAS Google Scholar
Acknowledgements
We thank Valerio Frazzini, Francesca Petrucci, and Mary Evangeline Oberschlake for the editing, figure preparation, and critical revision of the article. SLS was supported by funds from the Italian Department of Education (FIRB 2003, PRIN 2006, and PRIN 2008).
Author information
Author notes
- D Ciavardelli and E Silvestri: These authors contributed equally to this work.
Authors and Affiliations
- Molecular Neurology Unit, Center of Excellence on Aging (CeSI), University G. d'Annunzio, Chieti, Italy
D Ciavardelli, M Bomba, C Di Ilio & S L Sensi - Department of Biomedical Science, University ‘G. d'Annunzio’, Chieti-Pescara, Italy
D Ciavardelli & C Di Ilio - Department of Biological and Environmental Science, University of Sannio, Benevento, Italy
E Silvestri, A Del Viscovo, D De Gregorio, M Moreno, F Goglia & L M T Canzoniero - Department of Neuroscience and Imaging, University ‘G. d'Annunzio’, Chieti-Pescara, Italy
M Bomba & S L Sensi - Department of Neurology, University of California at Irvine, Irvine, CA, USA
S L Sensi
Authors
- D Ciavardelli
- E Silvestri
- A Del Viscovo
- M Bomba
- D De Gregorio
- M Moreno
- C Di Ilio
- F Goglia
- L M T Canzoniero
- S L Sensi
Corresponding authors
Correspondence toL M T Canzoniero or S L Sensi.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ciavardelli, D., Silvestri, E., Viscovo, A. et al. Alterations of brain and cerebellar proteomes linked to A_β_ and tau pathology in a female triple-transgenic murine model of Alzheimer's disease.Cell Death Dis 1, e90 (2010). https://doi.org/10.1038/cddis.2010.68
- Received: 10 June 2010
- Revised: 10 August 2010
- Accepted: 12 August 2010
- Published: 28 October 2010
- Issue Date: October 2010
- DOI: https://doi.org/10.1038/cddis.2010.68