Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis (original) (raw)
Main
Numerous clinical and experimental investigations have evaluated the role of inflammatory mediators in the development of alcoholic acute pancreatitis (AP). These factors have been usually determined in the serum, by total mRNA PCR or in isolated acinar cells in vitro.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 The origin of the pro- and anti-inflammatory cytokines and inflammasome-associated factors that are evident during the early onset of alcoholic AP has not been completely evaluated, and there is no adequate evidence that those inflammatory mediators colocalize with _α_-amylase-specific acinar cells in vivo.
Although NF_κ_B is the central transcription factor for pro-inflammatory factors such as TNF_α_, IL-6, MCP-1, and IL-18, and anti-apoptotic factors such as Bcl-XL, manipulation of the NF_κ_B pathway in acinar cell-specific transgenic mouse models has recently yielded contradictory results, suggesting caution in interpreting the role of NF_κ_B in models of pancreatitis and in clinical settings.11, 12, 13, 14, 15, 16, 17, 18 The NF_κ_B pathway is extremely complex as NF_κ_B-induced TNF_α_ expression is pro-inflammatory in many diseases, but can be pro-apoptotic or necrotic in pancreatitis, complicating the search for adequate therapeutic interventions for this potentially life-threatening conditions.19, 20
Experimental models of alcoholic pancreatitis have shown that lipopolysaccharide (LPS) exacerbates the development of alcoholic AP and that repeated exposure to LPS results in fibrogenesis and chronic pancreatitis (CP).21, 22, 23, 24 We recently reported that the combination of alcohol and endotoxemia attenuated the apoptotic response, and inhibited autophagy signaling promoted the early onset of AP, which supports clinical observations that episodes of acute and recurrent pancreatitis can progress to CP.22, 24 Alcoholics seem more susceptible to AP, a condition associated with an earlier onset, more frequent episodes and an increased risk of CP.25, 26, 27, 28 Alcoholic patients also have elevated serum LPS concentrations, which have been correlated with the severity of alcoholic AP.29, 30 Interestingly, chronic sub-clinical endotoxemia has been linked to insulin resistance, obesity, and diabetes, as well as cardiovascular disease, indicating that LPS has important clinical relevance not only for alcoholics with pancreatitis, but also for other metabolic disorders.31, 32
As various cell types within the pancreas and other organs may modulate inflammatory responses during the course of AP, we attempted to clarify the origin of the inflammatory mediators and the inflammasome components that are upregulated during the early onset of sub-clinical AP, both in human AP samples and in rats treated with alcohol and LPS. As the inflammatory response of acinar cells to endotoxin has become clinically relevant in alcoholism, we analyzed whether acinar cells respond directly to alcohol and LPS and are able to generate the injury signals necessary to initiate necro-inflammatory responses.
Results
Alcohol increases the LPS-induced inflammatory response in acinar cells
To understand the mechanisms by which chronic alcohol consumption and endotoxemia initiate pancreatic injury and AP, the well-established Lieber–DeCarli alcohol feeding model was used.33 The origin of inflammatory mediators was determined using double IF colocalization studies. TNF_α_, IL-6, MCP-1, and IL-10 expression were quantified alone and with α_-amylase colocalization in acinar cells. All cytokine levels were highly increased 3 h after LPS and colocalize with α_-amylase. Alcohol exposure significantly enhanced the levels of all evaluated cytokines 3 h after LPS. The cytokine levels returned to normal 24 h later (Figures 1a–f). A Luminex cytokine assay also confirmed a substantial increase in the levels of TNF_α, IL-6, MCP-1, and IL-10, 3 h after LPS stimulation in whole-tissue homogenate and serum samples, but there was no significant difference when comparing pair-fed controls and alcohol-fed rats 3 h after LPS exposure (Table 1). The inconsistency may be due to systemic interference by other organs such as the liver or due to the different LPS sensitivity of other cell types within the pancreas, such as islet or ductal cells. Real-time PCR for TNF_α and IL-6 confirm the elevated levels 3 h after LPS (Supplementary Figure 4).
Figure 1
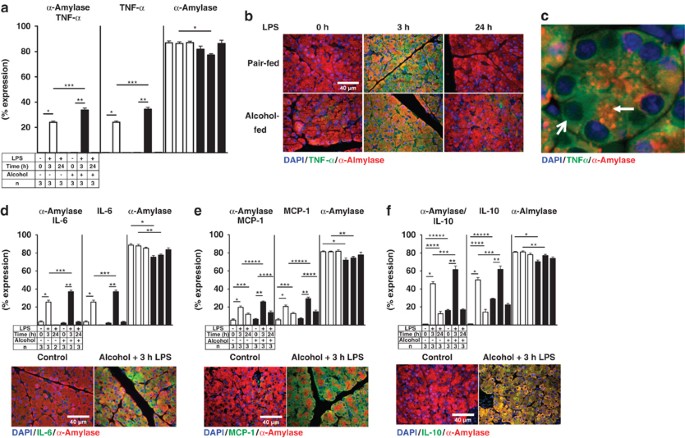
Alcohol and LPS treatment increased the expression of pro-, and anti-inflammatory mediator. (a) Ten randomly captured images of TNF_α_ and _α_-amylase were analyzed and scattergram values were plotted as means±S.E.M. for three animals per group. *_P_≤0.0001+170-fold; **_P_≤0.0001+165-fold; ***P_≤0.0001+1.4-fold. TNF_α alone: *_P_≤0.0001+109-fold; **_P_≤0.0001+170-fold; ***_P_≤0.0001+1.4-fold; _α_-amylase alone: *P_≤0.0001−1.15-fold. (b) Representative images of TNF_α (Cy2 green) and α_-amylase (Cy3 red). (c) Higher magnification of TNF_α (Cy2, green) and _α_-amylase (Cy3, red) acinar cell localization from alcohol-fed rats 24 h after LPS. α_-amylase is mainly expressed in the apical region of the acinar cell (arrow), whereas TNF_α is mainly expressed in the basolateral region of the acinar cells (open arrow). (d) Quantitation of IL-6 (Cy2, green) and _α_-amylase (Cy3, red). *_P_≤0.0001, +7.42-fold; **_P_≤0.001, +18-fold; ***_P_≤0.001, +1.44-fold. IL-6 alone: *_P_≤0.0001, +7.4-fold; **_P_≤0.001, +18-fold; ***_P_≤0.001, +1.42-fold. _α_-amylase alone: *_P_≤0.001, −1.17-fold; **_P_≤0.001, −1.13-fold. (e) Quantitation of MCP-1 (Cy2, green) and _α_-amylase (Cy3, red): *_P_≤0.0001, +3.6-fold; **_P_≤0.0001, both, +4-fold; ***_P_≤0.003, +2.2-fold; ****_P_≤0.016, +2.2-fold; *****_P_≤0.0018, +1.3-fold: MCP-1 alone: *_P_≤0.0001, +3.6-fold; **_P_≤0.0001, +4-fold; ***_P_≤0.027, +2.1-fold; ****_P_≤0.001, +2.2-fold; *****_P_≤0.0014, +1.41-fold: _α_-amylase alone: *_P_≤0.0001, −1.14-fold; **_P_≤0.0011, −1.09-fold. (f) Quantitation of IL-10 (Cy2, green) and _α_-amylase (Cy3, red): *_P_≤0.0001, +56-fold; **_P_≤0.0001, +3.8-fold; ***_P_≤0.0036, +1.3-fold; ****_P_≤0.0006, +14-fold; *****_P_≤0.0001, +20-fold. IL-10 alone: *_P_≤0.0001, +55-fold; **_P_≤0.0001, +2.1-fold; ***_P_≤0.0189, +1.22-fold; ****_P_≤0.0033, +15-fold; *****_P_≤0.0001, +31-fold. _α_-amylase alone: *_P_≤0.0001, −1.14-fold; **_P_≤0.0001, −1.05-fold
Table 1 Increased expression of inflammatory mediators in serum and whole tissue homogenate after LPS
A representative IF image for TNF_α_/α_-amylase is shown (Figure 1b). Quantitation of the expression of TNF_α, IL-6, MCP-1, and IL-10 was performed by scattergram analysis. A representative example is shown for TNF_α_/_α_-amylase in Supplementary Section (Supplementary Figure 1a). This result suggests that alcohol-injured acinar cells are capable of producing increased levels of inflammatory mediators in response to LPS. The expression of _α_-amylase alone was slightly reduced in the alcohol-fed animals (Figures 1a–f), suggesting that alcohol exposure alone influences _α_-amylase expression. In addition, all investigated inflammatory mediators were found in the basolateral region of the acinar cell, whereas α_-amylase was found mostly in the apical region, indicating a true distribution within acinar cells (representative shown for TNF_α/α_-amylase) (Figure 1c). The expression of TNF_α, IL-6, MCP-1, and IL-10 in all pancreatic cells showed an identical pattern as the acinar cell-specific _α_-amylase colocalization expression because only a few non-acinar cells were positive for those cytokines (Supplementary Table I). These findings indicated that both pro- and anti-inflammatory cytokines and chemokines were produced in acinar cells at the early onset of LPS-induced sub-clinical alcoholic pancreatic injury. Moreover, quantification of the overall acinar cell distribution within the rat pancreatic tissue revealed that 90% of the rat pancreas mass consisted of acinar cells (Supplementary Table I).
Intracellular translocation of LPS and cytokine expression in acinar-like AR42J cells is enhanced by alcohol in vitro
First, the translocation of labeled LPS was evaluated in acinar-like AR42J cells in vitro. Labeled LPS became internalized into AR42J cells positive for α_-amylase (Figure 2a). Similar to our results in vivo, LPS exposure increased the expression of TNF_α and the combination of alcohol and LPS also enhanced TNF_α_ expression compared with LPS alone. TNF_α_ colocalized with α_-amylase in acinar-like AR42J cells in vitro (Figure 2b). Moreover, silencing of the LPS translocation cascade using siRNA to target TLR-4 and MyD88 inhibited TNF_α expression in response to LPS (Figure 2c), indicating that LPS-induced production of inflammatory mediators was facilitated by TLR-4 signaling. A higher level of LPS-induced TNF_α_ expression was observed when LPS was administrated together with LBP and CD14, indicating a more efficient LPS response via TLR-4 signaling. The expression of other cytokines in response to LPS in AR42J cells was next determined in vitro. LPS exposure significantly increased _α_-amylase colocalization with IL-6 (Figure 2d) and MCP-1 (Figure 2e). These data indicate that LPS translocated into acinar-like AR42J cells, initiating an intracellular defense response by inducing the expression of pro-inflammatory mediators in vitro, consistent with our findings in vivo.
Figure 2
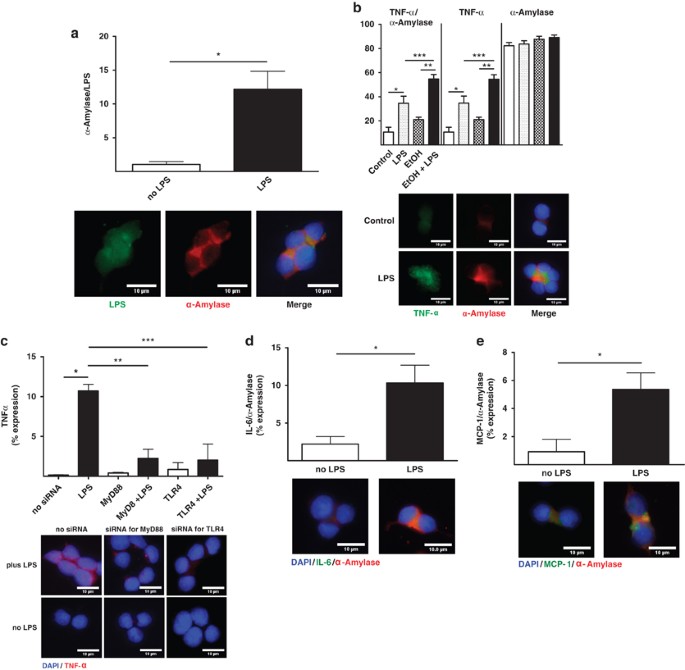
In vitro cytokines expression in response to alcohol and LPS. (a) LPS (Cy2 green) and _α_-amylase (Cy3 red) colocalization were plotted as means±S.E.M.: *P_≤0.01, +11-fold. (b) Quantitation of TNF_α (Cy3 green) and _α_-amylase (Cy5 red) colocalization values in response to alcohol and LPS were plotted as means±S.E.M.: *_P_≤0.01, +3.5-fold; **_P_≤0.0001, +2.6-fold; ***P_≤0.01, +1.6-fold. (c) TNF_α (Cy3 red) expression with or without MyD88 and TLR-4 siRNA. Expression values were plotted as means±S.E.M.: *_P_≤0.01;+72-fold; **_P_≤0.01, −4.7-fold by MyD88 siRNA plus LPS; ***_P_≤0.01, −5.2-fold by TLR-4 siRNA plus LPS. (d) IL-6 (Cy3 green) and _α_-amylase (Cy5 red) quantitation were plotted as means±S.E.M.: *_P_≤0.05,+4.6-fold. (e) MCP-1 (Cy3 green) and _α_-amylase (Cy5 red) quantitation were plotted as means±S.E.M.: *_P_≤0.001,+5.8-fold
Alcohol exacerbates LPS-induced inflammasome-associated expression of IL-18 and caspase-1 in acinar cells
The origin of the inflammasome-associated expression of IL-18 and caspase-1 was next determined using double IF colocalization studies. IL-18 and caspase-1 expression were quantified alone and with colocalization of acinar cell-specific _α_-amylase. Both IL-18 and caspase-1 were highly increased 24 h after LPS, colocalized with _α_-amylase and the expression was highly enhanced after alcohol exposure (Figures 3a and b). Identical results were obtained for IL-18 in a whole-tissue homogenate assay, while the serum levels of IL-18 showed a peak increase 3 h after LPS (Table 1), indicating that the systemic level of IL-18 may be influenced by other organs. This observation was confirmed by determining tissue caspase-1 activity. Caspase-1 activity was significantly increased 24 h after LPS, and LPS-induced caspase-1 activity was not affected by alcohol (Figure 3c). The expression of IL-18 and caspase-1 in all pancreatic cells was almost identical to their acinar cell-specific _α_-amylase colocalization because only a small number of non-acinar cells were positive for the inflammasome (Supplementary Table II). The expression of the inflammasome was next determined in response to LPS in AR42J cells in vitro. LPS exposure also significantly increased the colocalization of IL-18 and caspase-1 in vitro. We conclude that LPS is capable of initiating the inflammasome complex formation not only in vivo but also in vitro in response to LPS (Figure 3d). These results suggest that acinar cells respond to LPS in a manner that is similar to that reported for human monocytes and macrophages.34
Figure 3
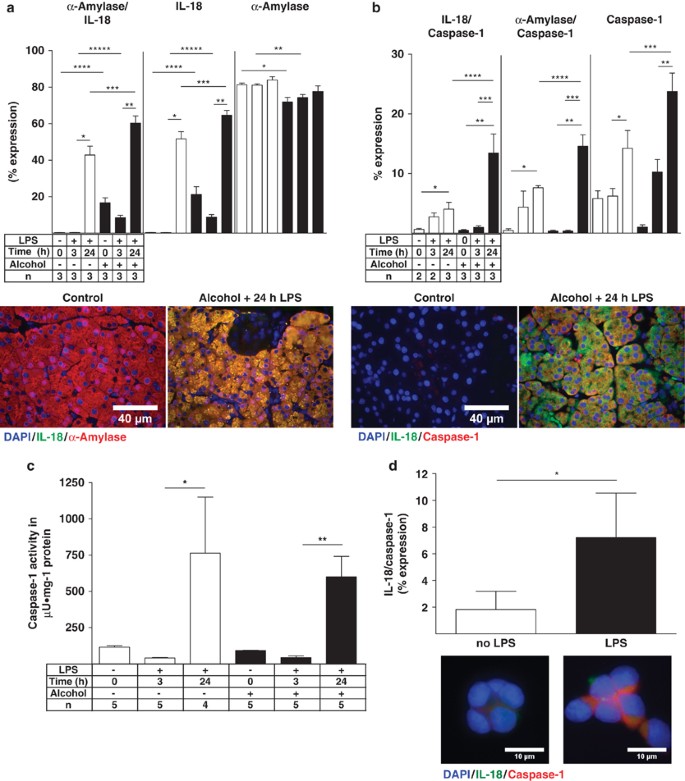
Pancreatic acinar cells expressing inflammasome-associated IL-18 and caspase-1. (a) Ten IL-18 (Cy2, green) and _α_-amylase (Cy3, red) randomly captured images were analyzed and plotted as means±S.E.M. for three animals per group. *_P_≤0.0001, +119-fold; **_P_≤0.0001, +7-fold; ***_P_≤0.012, +1.4-fold; ****_P_≤0.0001, +49-fold; *****_P_≤0.0001, +23-fold. IL-18 alone: *_P_≤0.0001, +159-fold; **_P_≤0.0001, +7.3-fold; ***_P_≤0.0278, +1.19-fold; ****_P_≤0.0001, +53-fold; *****_P_≤0.0001, +27-fold. _α_-amylase alone: *_P_≤0.0027, −1.13-fold; **_P_≤0.0011, −1.09-fold. (b) Quantitation of Caspase-1 (Cy2, green) and IL-18 (Cy3, red). *_P_≤0.0088, +6.5-fold; **_P_≤0.0011, +134-fold; ***_P_≤0.0128, +3.4-fold; ****_P_≤0.05, +3-fold. Caspase-1 alone: *_P_≤0.05, +2.3-fold; **_P_≤0.01, +2.3-fold; ***_P_≤0.05, +1.6-fold. Quantitation of caspase-1 and _α_-amylase. *_P_≤0.01, +19-fold; **_P_≤0.05, +40-fold; ***_P_≤0.05, +40-fold; ****_P_≤0.05, 1.9-fold. (c) Caspase-1 activity was plotted by means±S.E.M. for four to five animals per group. *_P_≤0.05, +19-fold; **_P_≤0.001, +14-fold. (d) In vitro Caspase-1 (Cy3 green) and IL-18 (Cy5 red) ‘inflammasome’ components colocalization quantitation in acinar-like AR42J cells were plotted as means±S.E.M.. *_P_≤0.05, +4-fold after LPS
Necro-inflammatory responses in a sub-clinical AP model
Pancreatic necrosis is believed to initiate inflammatory responses, fibrogenesis, and pancreatitis according to the necrosis–fibrosis hypothesis.35 Histopathological analysis showed mild edema and some vacuolization 24 h after LPS in both groups, as reported earlier.22, 23 No visible signs of intense acinar cell necrosis were observed in the tissues of pair-fed or alcohol-fed animals either 3 or 24 h after LPS administration (Figure 4a). We have previously shown that pancreatic HMGB1 is decreased in experimental alcoholic pancreatitis.22 We next extended our observation by evaluating nuclear HMGB1 in pancreatic acinar cells using the IF technique. Nuclear HMGB1 significantly decreased in the pair-fed group and in the alcohol-fed group 3 h after LPS, and returned to baseline levels 24 h later (Figure 4b). Serum HMGB1 and LDH increased in the pair-fed and alcohol-fed animals 3 h after LPS and returned to baseline 24 h later in both groups. Both tissue injury markers were higher in the alcohol-fed group compared with the pair-fed controls 3 h after LPS, although only LDH reached significance (Figures 4c and d). Our data show a loss of nuclear HMGB1 as early as 3 h after LPS and a subsequent return to normal levels, suggesting that acinar cells undergo sub-lethal necrotic injury during the early stages of LPS-induced sub-clinical pancreatitis and recover over the course of the disease. Infiltrating macrophages were next analyzed by ED2 staining, as described previously.36 Only a few ED2-positive macrophages were found in the controls, whereas ED2-positive macrophages increased only 2.5-fold in the alcohol-fed group 24 h after LPS (Figure 4e). Our results suggest infiltration by immune cells at the later stage of sub-clinical AP. Furthermore, the pancreatic acute-phase proteins pancreatitis-associated protein-1 (PAP-1) and pancreatic stone protein (PSP) increased ∼400-fold and 130-fold, respectively, in pair-fed and alcohol-fed animals 24 h after LPS, indicating the early induction of pancreatitis-associated acute-phase proteins (Supplementary Figures 2a and b).
Figure 4
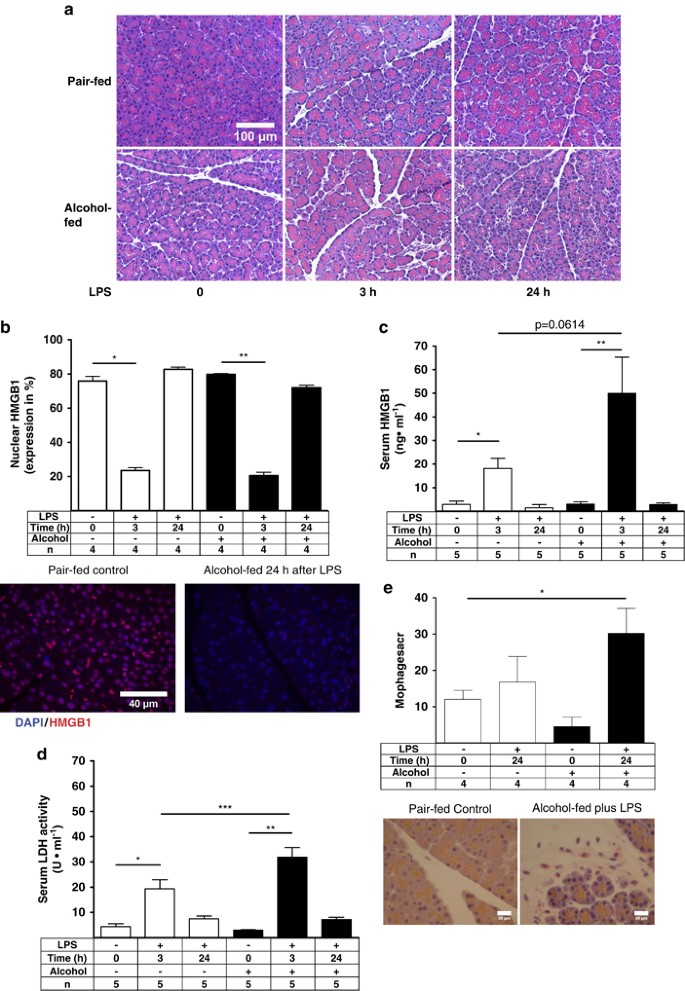
Alcohol and endotoxemia exacerbate pancreatic necrosis in sub-clinical model of AP. (a) Representative H&E-stained pancreatic tissue images with some edema and vacuolization. No visible necrosis is recognizable. (b) Nuclear HMGB1 (Cy5 red) values were plotted as means±S.E.M. for three to five animals per group. *_P_≤0.001, −2.3-fold; **_P_≤0.01, −2-fold. (c) Serum HMGB1 was determined using HMGB1 ELISA. Values were plotted as means±S.E.M. for five animals. *_P_≤0.05, +6.2-fold; **_P_≤0.01, +16-fold. (d) LDH values were plotted as means±S.E.M. for five animals. *_P_≤0.01, +4.4-fold; **_P_≤0.0001, +10-fold; ***_P_≤0.05, +1.6-fold. (e) Representative IHC images of pair-fed control and alcohol-fed rat tissue section 24 h after LPS stained for ED2 (CD 163)-positive macrophages using a light microscope with a × 40 objective. Plotted were means±S.E.M. for four animals per group. *_P_≤0.05, +2.4-fold
Human acinar cells expressing pro- and anti-inflammatory mediators in acute/recurrent pancreatitis specimens
Control tissue from donor (control) patients, CP patients, and acute/recurrent (AP/RAP) patients was examined to determine the expression of inflammatory mediators in areas of intact acini structure in hematoxilin and eosin (H&E)-stained tissue sections, in the same manner as described for the animal tissue. A representative image is shown in Figure 5a. TNF_α_, IL-1_β_, and IL-18 increased significantly in AP/RAP tissue compared with donor controls or CP tissue and colocalized with α_-amylase, suggesting that acinar cells produce the majority of these pro-inflammatory cytokines (Figures 5b–e). Similar to our finding in rats, inflammasome-associated caspase-1 and IL-18 also increased substantially and caspase-1 colocalized α_-amylase in AP/RAP tissue compared with donor controls or CP tissue, indicating that inflammasome-associated IL-18/caspase-1 expression is evident in acinar cells from AP/RAP patients (Figure 5e). TNF-α and inflammasome components were also slightly increased in CP samples compared with donor controls, but the increase was significantly less pronounced than in AP/RAP tissue (Figures 5b–e). IL-6 and IL-10 expression also increased substantially in AP/RAP samples compared with donor controls and CP patients (Figure 5d), indicating pro- and anti-inflammatory cytokine production in human acute, recurrent pancreatitis specimens. Quantitation of the expression levels of TNF_α, IL-1_β, IL-6, IL-10, IL-18, and caspase-1 was performed by scattergram analysis. A representative example for TNF-α and its colocalization with α_-amylase is shown (Supplementary Figure 1b). These data were confirmed by counting all evaluable cells in 10 images for TNF_α, IL-1_β_, and IL-18/caspase-1 positivity, alone or colocalized with α_-amylase. Only few α_-amylase-negative cells were positive for TNF_α, IL-1_β, or IL-18/caspase-1, indicating that the majority of pro- and anti-inflammatory mediators were produced by pancreatic acinar cells in acute/recurrent pancreatitis (Supplementary Table III). Our experimental observation was therefore confirmed in clinical specimens. These findings suggest that CP can be established without acinar cell necrosis and that pancreatic acinar cells are the first responders in driving inflammation by expressing inflammatory mediators.
Figure 5
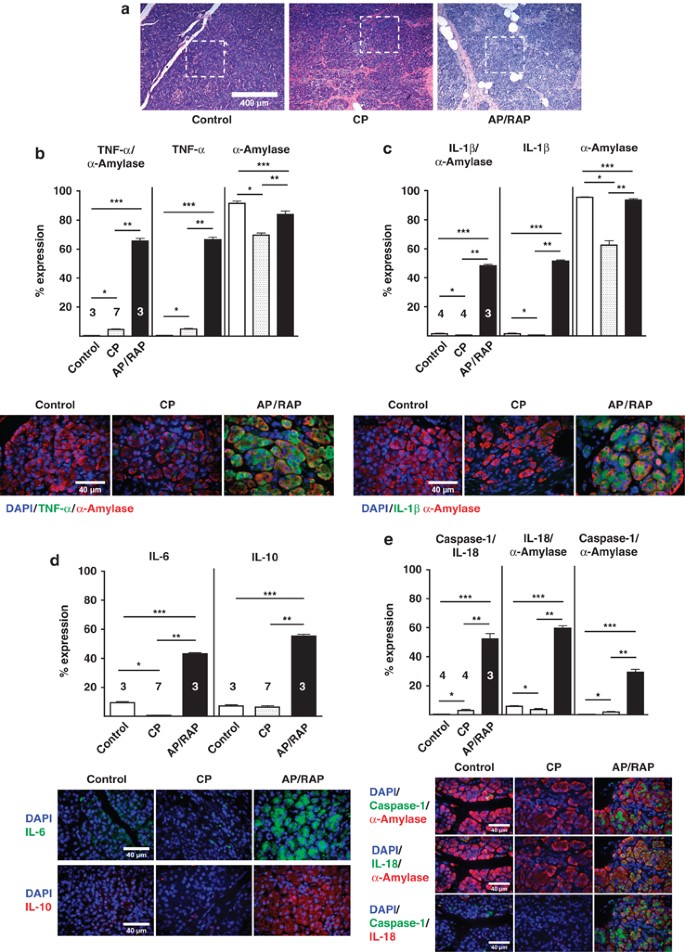
Elevated expression of inflammatory mediators in acinar cells from human AP specimens. (a) Representative H&E-stained tissue section images indicating areas of intact acini structures of healthy controls, CP and acute/recurrent pancreatitis samples, were selected for the IF quantitation. (b) Representative IF images for TNF_α_ (Cy3 green) and _α_-amylase (Cy5 red) and quantitation levels were plotted as means±S.E.M. for three to seven human specimens. *_P_≤0.0001, +11-fold; **_P_≤0.0001, +14-fold; ***P_≤0.0001, +170-fold. TNF_α alone: *_P_≤0.0001, +12-fold; **_P_≤0.0001, +13-fold; ***_P_≤0.0001, +153-fold. _α_-amylase alone: *_P_≤0.0001, −1.3-fold; **_P_≤0.0001, −1.2-fold; ***P_≤0.0135, −1.1-fold. Identical to rat acinar cells, human TNF_α is also mainly expressed in the basolateral region and α_-amylase in the apical region of the acinar cell. (c) Quantitation IL-1_β (Cy3 green) and _α_-amylase (Cy5 red) levels were plotted as means±S.E.M. for 3–4 human specimens. *_P_≤0.031, −3-fold; **_P_≤0.0001, +106-fold; ***P_≤0.0001, +34-fold. IL-1_β alone: *_P_≤0.0234, −3-fold; **_P_≤0.0001, +106-fold; ***_P_≤0.0001, +34-fold. _α_-amylase alone:*_P_≤0.05, −3-fold; **_P_≤0.01, −1.34-fold; ***_P_≤0.01, −1.08-fold (d) Quantitation of IL-6 (Cy3 green) levels were plotted as means±S.E.M. for three to seven human specimens. *_P_≤0.0001, −16-fold; **_P_≤0.0001, +78-fold; ***_P_≤0.0001, +4.6-fold. Similar, IL-10 (Cy5 red) levels were plotted as means±S.E.M. for three to seven human specimens. **_P_≤0.0001, +8.7-fold; ***_P_≤0.0001, +7.7-fold. Note, both IL-6 and IL-10 were also mainly expressed in the basolateral region of the acinar cell. (e) Quantitation of IL-18 (Cy5 green), caspase-1 (Cy2 red) values were plotted as means±S.E.M. for three to four human specimens. *_P_≤0.01, +7.3-fold; **_P_≤0.0001, +17-fold; ***_P_≤0.0001, +130-fold. IL-18 (green) and _α_-amylase (Cy3 red) quantitation levels were plotted as means±S.E.M. for three to four human specimens. *_P_≤0.05, −1.5-fold; **_P_≤0.0001, +16-fold; ***_P_≤0.0001, +10-fold. Caspase-1 (green) and _α_-amylase (Cy3 red) quantitation levels were plotted as means±S.E.M. for three to four human specimens. *_P_≤0.0001, +107-fold; **_P_≤0.001, +7.4-fold; ***_P_≤0.0001, +14.5-fold
Human and rat acinar cells express TLR-4
Although TLR-4 has recently been shown to be involved in pancreatitis,37, 38 the expression of TLR-4 in pancreatic acinar cells has not been demonstrated. Therefore, TLR-4 and _α_-amylase colocalization was evaluated in our rat and human pancreatitis samples. TLR-4 expression was evident in rat pancreatic acini (Supplementary Figure 3a). This result suggests that LPS can mediate the inflammatory response through TLR-4 in vitro and in experimental sub-clinical pancreatitis. Furthermore, TLR-4 also colocalize with labeled LPS in AR42J cells in vitro (Supplementary Figure 5a). In addition, acute/recurrent human pancreatitis specimens also show increased expression of TLR-4, presumably independently of LPS signaling. Our data indicate that the expression of pro-inflammatory mediators in acute or recurrent pancreatitis is due to the involvement of TLR-4 in pancreatic acini (Supplementary Figure 3b).
Discussion
Our findings indicate that pancreatic acinar cells can produce cytokines and chemokines that are involved in the inflammatory response, including the inflammasome-associated factors IL-18 and caspase-1, which are found in the basolateral region of acinar cells. The expression of all inflammatory mediators was highly enhanced in response to LPS in alcohol-fed rats, indicating that chronic alcohol exposure exacerbated acinar cell-specific inflammatory responses in this model of sub-clinical AP. The expression profile of cytokines and chemokines, as well as the inflammasome-associated expression of IL-18 and caspase-1, clearly indicate that the inflammatory mediators released during the early response to LPS are produced exclusively by acinar cells, because few α_-amylase-negative cells were positive for these factors. In addition, alcohol-injured acinar cells increase their expression of inflammatory mediators and inflammasome-related components after endotoxemia, suggesting that the alcohol-exacerbated LPS response that initiates sub-clinical AP is mediated by acinar cells. Our systemic serum and whole-tissue homogenate assay for validating the expression of TNF_α, IL-6, IL-10, MCP-1, and IL-18 confirm the substantial increase in these factors after LPS treatment but did not show an additional increase in expression after alcohol exposure, indicating that systemic serum and whole-tissue homogenate approaches are inadequate for investigating local inflammatory responses in pancreatitis.
Samples obtained from patients with pancreatitis also revealed that acinar cells were responsible for the production of TNF_α_, IL-1_β_, IL-6, IL-10, and inflammasome IL-18/caspase-1, indicating that pancreatic acinar cells are the major source of pro- and anti-inflammatory cytokines and the inflammasome components in patients with AP/RAP. Our findings are supported by a recent experimental study of cerulein-induced AP, in which NF_κ_B expression was detected in acinar cells.14, 17 Thus, acinar cells are the major source of inflammatory mediators after early pancreatic injury and during the early onset of sub-clinical AP, with these responses occurring via the TLR-4 signaling pathway.
LPS was internalized by α_-amylase-positive acinar-like AR42J cells via TLR-4 signaling. Moreover, LPS-induced TNF_α production was enhanced by alcohol exposure, and TNF_α_ production was suppressed by the silencing of TLR-4 and MyD88 signaling in vitro, indicating that LPS drives the production of inflammatory mediators through the TLR-4 receptor. Human and rat pancreatic acinar cells express TLR-4, indicating that acinar cells are capable of responding directly to LPS. Earlier investigations have reported that TLR-4 signaling is also involved in the inflammatory responses in experimental AP independent from LPS, and TLR-4 deficiency diminished the severity of cerulein-induced AP.37, 39, 40, 41 However, it remains unclear how cerulein facilitates TLR-4 dependent inflammatory responses in AP. To our knowledge, these results are the first to show that the response of pancreatic acinar cells to LPS is similar to the reported response of human monocytes and macrophages.34 Moreover, these results are the first to show that pancreatic acinar cells express both pro- and anti-inflammatory cytokines and chemokines, including inflammasome components, and participate in the initiation of AP-associated inflammatory signals. Pancreatic acute-phase proteins PAP-1 and PSP were substantially increased by LPS treatment and treatment and were further increased by alcohol exposure, although this did not reach statistical significance. Elevated serum IL-18 concentrations have been found to correlate with the clinical severity of AP, and persistently elevated IL-18 expression has been associated with a reduced ability to recover from pancreatic injury in alcoholics.2, 42, 43, 44 This is in agreement with results showing that a caspase-1 inhibitor can reduce the severity of experimental AP.45 Enhanced expression of inflammasome-associated IL-18 and caspase-1 may therefore contribute to more severe acinar cell injury in alcoholic pancreatitis patients.
Tissue release of the nuclear protein HMGB1 correlated well with the systemic necrosis markers LDH and HMGB1, indicating that loss of nuclear HMGB1 was an early event in necrotic acinar cells. Most alcohol- and LPS-injured acinar cells apparently undergo some degree of necrotic cell death. However, nuclear HMGB1 normalized rapidly, suggesting that its levels were restored.
MCP-1 expression was also elevated in acinar cells exposed to alcohol and LPS, suggesting that acinar cells can initiate inflammatory responses in the absence of other pancreatic cell types. Indeed, acinar cells were able to produce high levels of MCP-1 during the early stages of sub-clinical AP and were therefore involved in subsequent (MCP-1-driven) monocyte chemotaxis and immune cell infiltration.46, 47 By contrast, the expression of the anti-inflammatory cytokine IL-10 was markedly increased 3 h after LPS exposure in alcohol-treated rats, indicating that IL-10 contributes to the regulation of pancreatic pro-inflammatory responses and may participate in the resolution phase. As IL-10 reduces the severity of AP, it may also attenuate AP severity in our model.48
Animal models are critical for evaluating early tissue responses to pancreatic injury, because pancreatic tissue is not available from human alcoholic AP/RAP patients. However, acinar cell expression of inflammasome components and necro-inflammatory cytokines was confirmed in human AP/RAP tissue specimens. Detection of TNF_α_, IL-1_β_, IL-6, IL-10, IL-18, and caspase-1 expression in intact acinar cells in patients with AP and in a rat model, in the absence of visible areas of necrotic tissue, indicates that CP can be established from sub-clinical alcoholic AP with little acinar cell necrosis. Acinar cells respond to injury-associated stimuli, such as endotoxin, by producing pro- and anti-inflammatory cytokines, including the inflammasome components, similar to human macrophages and monocytes.34
This investigation provided evidence that pancreatic acinar cells are capable of producing pro- and anti-inflammatory cytokines, including those that are associated with the inflammasome and those that induce monocyte chemotaxis, in clinical AP/RAP samples and in an experimental animal model of AP in response to LPS. Furthermore, alcohol was capable of exacerbating acinar cell-mediated inflammatory responses.
Materials and Methods
Animals and reagents
Male Sprague-Dawley rats were purchased from Charles River (Sulzfeld, Germany). All experiments were approved by the Institutional Animal Care and Use Committee in accordance with the guidelines of the University of Heidelberg and the Federal Presiding Board of Animal Care Karlsruhe, Germany. Antibodies were selected according to proven functionality in formalin-fixed paraffin-embedded (FFPE) tissue sections by the seller or by publication records. Mouse monoclonal anti-α_-amylase (sc-46657), anti-HMGB1 (sc-56698) as well as goat polyclonal anti-IL-18 (sc-6177), anti-IL-10 (sc-1783), and rabbit polyclonal anti-MCP-1 (sc-28879) were purchased from Santa Cruz Biotech (Heidelberg, Germany). Rabbit polyclonal anti-TNF_α (ab66579), anti-IL-6 (ab6672), anti-TLR-4 (ab13556) and anti-caspase-1 (ab1872) were purchased from Abcam (Cambridge, UK). Secondary anti-rabbit Cy3 or Cy5-conjugated, anti-goat-Cy2-conjugated and anti-mouse Cy5-conjugated were obtained from Medac (Medac GmbH, Wedel, Germany). Fluorometric caspase-1 substrate (Ac-WEHD-AMC) and caspase-1 (Ac-WEHD-Al) inhibitor were purchased from Bachem (Bubendorf, Switzerland). HMGB1 ELISA was purchased from Shino-Test Corp. (Shino-Test, Tokyo, Japan). Luminex rat cytokine/chemokine multiplex panel was purchased from Linco (Saint Charles, MO, USA). RNA interference targeting TLR-4 and MyD88 mRNA were obtained from Qiagen as premade FlexiTube containing 4 different siRNAs for each target, to ensure TLR-4 and MyD88 mRNA suppression (Qiagen, Hilden, Germany). Serum LDH was determined by our institutional blood analysis center. All other chemicals were from Sigma Aldrich (Deisenhofen, Germany), if not stated otherwise.
Experimental design
Male rats weighing 150–175 g were fed a commercially available liquid diet (Lieber-DeCarli,33 BioServ, Frenchtown, NJ, USA) containing either ethanol (6% final concentration, equivalent to 36% of caloric intake in 15 rats) or maltose-dextrin (pair-fed group in 15 rats) in isocaloric amounts as described previously.22 After 14 weeks of feeding the control or alcohol diet, gram-negative bacterial LPS (E. coli, 026-B6; 0.8 mg/kg body weight, i.v.) or vehicle control (sterile saline) were injected once intravenously. Rats were divided into six groups each with five animals per group: pair-fed (PF) (_n_=5) and alcohol-fed controls (EtOH) (_n_=5), and pair-fed or alcohol-fed animals injected with LPS, sedated at 3 or 24 h after LPS injection (_n_=5) with pentobarbital (Nembutal, 60 mg/kg body weight, i.p.). The whole pancreas was resected, immersed in liquid nitrogen and stored at −80 C. Serum samples were also taken und stored at −80 C.
Human subjects
The study was approved and renewed in August 2012 by the Ethics Committee of the University Medical Faculty of Heidelberg. Human pancreata were obtained from three healthy donors, three acute/recurrent and up to seven CP patients attending the Department of Surgery of our University. In detail, the series comprised three cases of AP/RAP, were obtained from three men at the ages of 60, 64, and 70 years (average 64 years). Alcohol consumption was known in one of these patients. The remaining two patients did not have a reported history of alcohol, and pancreatitis occurred after low cardiac output syndrome following heart transplantation and unknown idiopathic reasons in one case each. The CP group consisted of five male and two female patients at ages between 52 years and 70 years (average 58 years). A history of alcoholism was known in five of these patients, one case represented an obstructive pancreatitis due to IMPN-related pancreatic cancer, and in one patient the CP was idiopathic. The indications for pancreatic head resections were pain and CP-related complications in organs neighboring the pancreas, such as common bile duct stenosis, main pancreatic duct stenosis, vascular obstruction, and/or duodenal obstruction. Normal human pancreatic tissues were obtained from one female and two male patients (ages of 31, 59 and 72 years, average 54 years) who were free of pancreatic disease (through an organ donor program in which there were no candidates for pancreatic transplantation).
Freshly removed tissue samples were immediately fixed in 4% buffered formalin. Microscopically, the control samples revealed regular pancreatic tissue, consisting of lobules of acinar cells, ducts, and scattered islets. Focally, a mild fibrosis was detected. However, there was no significant inflammation. In CP, the pancreatic tissues displayed chronic inflammation with scattered lymphocyts. Furthermore, peri-ductal, interlobular, and intra-lobular fibrosis was seen in various extents without necrosis. In acute/recurrent pancreatitis, the samples showed multiple areas of fatty necrosis and infiltrates of neutrophils. Two of the latter cases displayed pre-existing CP with fibrosis.
Immunofluorescence
IF was conducted using 7-μ_m thin pancreata FFPE tissue sections obtained from rat and human specimens and processed as described.22 Primary antibodies were incubated in antibody diluent (Dako, Hamburg, Germany) for 30 min at 37 °C in a dark humidified chamber, followed by several steps of washing. Sections were then incubated with anti-mouse Cy5 and/or anti-rabbit Cy3 and/or goat-Cy2 labeled secondary antibodies for 30 min at RT, also followed by several steps of washing and incubation with DAPI for 20 min. The sections were mounted in Fluoromount-G Reagent (Southern Biotech, Birmingham, AL, USA). Up to 10 images per slide/sample with typically 750–2250 cells ( × 63 objective) were captured using a Zeiss Axiovert 200 M microscope (Zeiss, Göttingen, Germany) equipped with DAPI, Cy2, Cy3 and Cy5 filters (Chroma Technology, Rockingham, VT, USA), as described recently.22 Briefly, standardized automatic acquisition was performed by the TissueFACS software (TissueGnostics, Vienna, Austria) controlling filters, exposure, camera (PCO, Kehlheim, Germany), and motor stage (Märzhäuser, Wetzlar, Germany). All images were processed using the TissueQuest software (TissueGnostics), allowing the quantitation of the total cell numbers from DAPI positive cells, as well as quantitation of target-positive cells. In FACS-like scatter-grams, the cells were plotted according to their Cy2, Cy3, and Cy5 IF intensity versus their DAPI-intensity from 10 randomly captured images. IF-positive cells were gated in the scatter-grams according to negative controls (no primary antibody), and the fluorescence intensity was expressed as percentage of the intensity of the DAPI staining, as described recently.22 Likewise, cellular colocalization of rat and human α_-amylase with TNF_α, IL-1_β, IL-6, IL-10, IL-18, and MCP-1 were analyzed in the same way with the software supported colocaliation analysis.
In vitro LPS and alcohol stimulation
Rat pancreatic acinar-like AR42J cell line was purchased from American Type Culture Collection (Manassas, VA, USA) and were grown in Ham’s F-12 medium supplemented with 20% heat-inactivated fetal calf serum (FCS), 50 U/ml penicillin G, 50 _μ_g/ml streptomycin, and 2.5 mg/ml plasmocin. The cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Cells were suspended and plated on ibidi _μ_-slide (ibidi GmbH, Martinsried, Germany) at a density of 12 000 cells per well and incubated for 24 h. They were exposed to starvation for another 24 h followed by incubating with either 10 _μ_g/ml fluorescence-conjugated LPS-Alexa488 (Cy2) (Molecular Probes, Life Technologies, Thermo Scientific, Carlsbad, CA, USA) or unlabeled LPS, as well as 1.2 _μ_g/ml per well recombinant CD14 and LBP (Abnova, Heidelberg, Germany), for an additional 16 h. After fixation with 100% ice-cold acetone, cells were IF-stained for cytokine expression, as described earlier.22
For the alcohol and LPS experiments, AR42J cells were suspended and plated onto sterile coverslips (Nalgene, Thermo Scientific, Waltham, MA, USA) in six-well tissue culture plate at a density of 300 000 cells per well and incubated for 24 h. The cells were treated with 250 mM ethanol, with or without 10 μ_g/ml LPS for another 24 h. After fixation with 100% ice-cold acetone for 30 min in dark at RT, the cells were IF-stained for TNF_α and _α_-amylase, as described earlier. All cells were mounted in Flauoromount-G Reagent (SouthernBiotech) and a minimum of five images per treatment were captured and analyzed using the same microscope and software as described.22 All in vitro experiments were at least four times repeated.
TLR-4 and MyD88 silencing
We and others have previously shown that LPS can induce pancreatic damage and pancreatitis.21, 22 It remains unknown how LPS exposure mediates acinar cell injury in the pancreas. We therefore determined LPS responsiveness in acinar-like cells in vitro and whether LPS internalized via Toll-like Receptor-4 and whether LPS-induced inflammatory mediators are dependent on intracellular adapter molecule MyD88, facilitating pro- and anti-inflammatory cytokine expression. Twelve-thousand AR42J cells were grown on ibidi eight-well _μ_-slides for 24 h followed by RNA interference for TLR-4 and MyD88. Transfection was accomplished using rat-specific TLR-4 and MyD88 small interfering RNA (siRNA), control siRNA, and HyPerFect transfection reagent, according to the instructions (Qiagen). Twenty-four hours after the transfection, the cells were exposed to LPS 10 _μ_g/ml, as well as 1.2 μ_g/ml per well recombinant CD14 and LBP (Abnova), for an additional 16 h. After fixation in 100% ice-cold acetone, cells were stained with primary anti-rat TNF_α (rabbit) and labeled secondary anti-rabbit Cy2 antibody, as well as DAPI staining, as described.22 Cells were mounted in Fluoromount-G Reagent (SouthernBiotech) and images were captured using the same microscope and software, as described.
Pancreatic histology
Pancreata were cut into 4-_μ_m-thick serial sections from 4% FFPE blocks. Sections were stained with Hematoxylin/Eosin, and examined by a pathologist who was not aware of the sample identity (FB). Tissue sections (_n_=4) were examined for parenchymal edema, acinar cell vacuolization, necrosis, inflammatory cell infiltration, and hemorrhage and analyzed in 10 randomly selected fields with the aid of the Olympus BX40 microscope camera system, as described previously.22
Immunohistochemistry
ED2 (CD 163) IHC was accomplished in paraffin-embedded pancreatic sections as described previously. ED2-positive cells, representative of mature macrophages, were examined and analyzed (_n_=5) in 20 randomly selected fields with the aid of a Zeiss Axioplan2 Imaging microscope (Zeiss, Feldbach, Germany).36
Caspase-1 activity
Activity of caspase-1 (_n_=4 to 5) was measured in pancreatic tissue extracts using a modified assay for measuring caspase activity as described previously.23 Briefly, the assay was performed according to the manufacturer’s instructions (Sigma Chemicals) with one exception: the assay was performed at 37 °C and not at room temperature, in order to imitate body temperature. Caspase-1 substrate (1.66 _μ_l/ml 10 mM Ac-WEHD-AMC) was incubated with pancreatic tissue extracts with or without Caspase-1 inhibitor (Ac-WEHD-Al). Fluorescence was measured using a fluorometic spectrometer Synergy HT (Bio-Tek Inc., Winooski, VE, USA). Results were expressed as the specific caspase-1 substrate cleavage per mg total protein per min, after subtraction of the nonspecific product formation (substrate plus inhibitor).
PAP-1 and PSP ELISA
Pancreatic secretory stress proteins PAP-1 and PSP are highly elevated in pancreatitis and effective marker for human and experimental pancreatitis.49 Pancreatic tissue extract was diluted in PBS and analyzed by highly specific PAP-1 and PSP ELISA, kindly analyzed by Professor Graf’s laboratory as described previously.49
Luminex multiplex analysis for cytokines and chemokines
Luminex cytokine multiplex analysis was performed in order to evaluate variations in the expression of cytokines and chemokines in our serum and tissue samples. Rat frozen tissues were homogenized on ice as described previously.22 Four animals of each group (n_=4) were analyzed according to the instruction using serum and tissue extract (Lincoplext rat cytokine kit (LINCO Research, Inc.). Interleukin (IL)-1_β, IL-6, IL-10, IL-18, TNF_α_ and MCP-1 were simultaneously quantified as described previously.50
Real-time PCR
Frozen whole pancreatic tissue was powdered in the presence of liquid nitrogen and extracted using TRIzol-Reagent (Life Technologies, Invitrogen, Thermo Scientific). Five micrograms of total RNA was reverse-transcribed using SuperScript II (Life Technologies, Invitrogen) according to the manufacturer’s instructions. Real-time PCR was analyzed on a TaqMan 7000 (Applied Biosystems, Foster City, CA, USA) under standard conditions, using either the TaqMan or SYBR-Green system. The calculated cycles were subtracted from the 18 s RNA (Applied Biosystems) reference, which was always included on the same plate. The difference in cycles was then calculated and expressed as relative transcript levels (2ΔΔ). The primer sequence for IL-6: forward GCCCTTCAGGAACAGCTATGA, reverse TCCGCAAGAGACTTC and FAM CATCAGTCC CAAGAAGGCAACT. TNF_α_: forward GCTCCCTCTCATCAGTTCCAT, reverse CCTCACACT CAGATCAT and FAM GGCTTGTCACTCGAGTTTTGAGAA. Real-time PCR for TNF_α_ and IL-6 were performed as described recently.51
Pancreatic tissue nuclear fractionation for HMGB1 ELISA
Pancreatic tissue (_n_=5) was fractionated according to a modified protocol, as previously described.52 Briefly, pancreatic frozen tissue was disrupted at 4 °C by a Teflon homogenizer in an extraction buffer containing 250 mM sucrose, 20 mM HEPES-KOH (pH 7.0), 10 mM KCl, 1 mM EGTA, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 mM PMSF and protease inhibitor mixture (1 _μ_g/ml each of pepstatin, leupeptin, and L-leucinethiol), and allowed to swell for 30 min at 4 °C. Homogenates were centrifuged (1000 × g, 10 min, 4 °C) to pellet nuclei and cell debris. The supernatants were centrifuged (13 000 × g, 30 min, 4 °C) and the cytosolic fractions (supernatant) were collected. The pellets (heavy membranes enriched with mitochondria) were resuspended in lysis buffer containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulphonyl fluoride, leupeptin, pepstatin and L-leucinethiol, each at 1 _μ_g/ml, and 0.1% Nonidet P-40 pH 7.9, then, aliquoted and stored at −80 °C. Protein concentrations in nuclear, cytosolic and mitochondrial fractions were determined by the BCA protein assay procedure (Pierce, Rockford, IL, USA). Nuclear extract and serum samples were analyzed by using HMGB1 ELISA kit II according to the instruction (Shino-Test Corporation).
Statistical analysis
Statistical analysis was performed by using the ANOVA, followed by Student’s _t_-test for each experimental group treated with LPS or vehicle. Results were considered significant when the _P-_value ≤0.05, indicated with * and were reported as mean±S.E.M. (standard error of the mean) or as normalization to control samples as indicated in the figure legend. Statistical calculations were performed using software from GraphPad Prism 5 (La Jolla, CA, USA).
Abbreviations
AMC:
7-amido-4-methylcoumarin
AP:
acute pancreatitis
CP:
chronic pancreatitis
ED2:
CD 163
EtOH:
alcohol-fed
H&E:
hematoxilin and eosin staining
IHC:
immunohistochemistry
IF:
immunofluoresence
HMGB1:
high mobility group box 1
(IL)-6:
interleukin-6
LPS:
lipopolysaccharide
LDH:
lactate-dehydrogenase
MyD88:
myeloid differentiation primary response gene 88
MCP-1:
monocyte chemo attractant protein-1
NF_κ_B:
nuclear factor _κ_B
PF:
pair-fed
PCR:
polymerase chain reaction
RFU:
release fluorescence unit
RAP:
recurrent acute pancreatitis
TNF_α_:
tumor necrosis factor-α
TLR-4:
Toll-like Receptor-4
References
- Mayer J, Rau B, Gansauge F, Beger HG . Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut 2000; 47: 546–552.
Article CAS Google Scholar - Pastor CM, Morel DR, Vonlaufen A, Schiffer E, Lescuyer P, Frossard JL . Delayed production of IL-18 in lungs and pancreas of rats with acute pancreatitis. Pancreatology 2010; 10: 752–757.
Article CAS Google Scholar - Brivet FG, Emilie D, Galanaud P . Pro- and anti-inflammatory cytokines during acute severe pancreatitis: an early and sustained response, although unpredictable of death. Parisian Study Group on Acute Pancreatitis. Crit Care Med 1999; 27: 749–755.
Article CAS Google Scholar - Ramudo L, Manso MA, Sevillano S, de Dios I . Kinetic study of TNF-alpha production and its regulatory mechanisms in acinar cells during acute pancreatitis induced by bile-pancreatic duct obstruction. J Pathol 2005; 206: 9–16.
Article CAS Google Scholar - Gukovskaya AS, Mareninova OA, Odinokova IV, Sung KF, Lugea A, Fischer L et al. Cell death in pancreatitis: effects of alcohol. J Gastroenterol Hepatol 2006; 21 (Suppl 3): S10–S13.
Article CAS Google Scholar - Brunner R, Xie J, Bank S . Does acute alcoholic pancreatitis exist with preexisting chronic pancreatitis? J Clin Gastroenterol 2004; 38: 201–202.
Article Google Scholar - Schneider L, Hackert T, Longerich T, Hartwig W, Fritz S, Krych R et al. Effects of gadolinium chloride and glycine on hepatic and pancreatic tissue damage in alcoholic pancreatitis. Pancreas 2010; 39: 502–509.
Article CAS Google Scholar - Panek J, Kusnierz-Cabala B, Dolecki M, Pietron J . Serum proinflammatory cytokine levels and white blood cell differential count in patients with different degrees of severity of acute alcoholic pancreatitis. Polski przeglad chirurgiczny 2012; 84: 230–237.
Article Google Scholar - Novovic S, Andersen AM, Ersboll AK, Nielsen OH, Jorgensen LN, Hansen MB . Proinflammatory cytokines in alcohol or gallstone induced acute pancreatitis. A prospective study. Jop 2009; 10: 256–262.
PubMed Google Scholar - Szabo G, Mandrekar P, Oak S, Mayerle J . Effect of ethanol on inflammatory responses. Implications for pancreatitis. Pancreatology 2007; 7: 115–123.
Article CAS Google Scholar - Algul H, Treiber M, Lesina M, Nakhai H, Saur D, Geisler F et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest 2007; 117: 1490–1501.
Article Google Scholar - Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD . NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 2002; 122: 448–457.
Article CAS Google Scholar - Dunn JA, Li C, Ha T, Kao RL, Browder W . Therapeutic modification of nuclear factor kappa B binding activity and tumor necrosis factor-alpha gene expression during acute biliary pancreatitis. Am Surgeon 1997; 63: 1036–1043 Discussion 1043–1034.
CAS PubMed Google Scholar - Huang H, Liu Y, Daniluk J, Gaiser S, Chu J, Wang H et al. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 2012; 144: 202–210.
Article Google Scholar - Satoh A, Shimosegawa T, Fujita M, Kimura K, Masamune A, Koizumi M et al. Inhibition of nuclear factor-kappaB activation improves the survival of rats with taurocholate pancreatitis. Gut 1999; 44: 253–258.
Article CAS Google Scholar - Steinle AU, Weidenbach H, Wagner M, Adler G, Schmid RM . NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999; 116: 420–430.
Article CAS Google Scholar - Treiber M, Neuhofer P, Anetsberger E, Einwachter H, Lesina M, Rickmann M et al. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology 2012; 141: 1473–1485 1485 e1471–1477.
Article Google Scholar - Gukovsky I, Gukovskaya AS . Nuclear factor-_κ_B in pancreatitis: jack-of-all-trades, but which one is more important? Gastroenterology 2012; 144: 26–29.
Article Google Scholar - Sendler M, Dummer A, Weiss FU, Kruger B, Wartmann T, Scharffetter-Kochanek K et al. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 2013; 62: 430–439.
Article CAS Google Scholar - Radhakrishnan SK, Kamalakaran S . Pro-apoptotic role of NF-kappaB: implications for cancer therapy. Biochim Biophys Acta 2006; 1766: 53–62.
CAS PubMed Google Scholar - Vonlaufen A, Xu Z, Daniel B, Kumar RK, Pirola R, Wilson J et al. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology 2007; 133: 1293–1303.
Article CAS Google Scholar - Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 2009; 137: 350–360 360 e351–e355.
Article CAS Google Scholar - Fortunato F, Deng X, Gates LK, McClain CJ, Bimmler D, Graf R et al. Pancreatic response to endotoxin after chronic alcohol exposure: switch from apoptosis to necrosis? Am J Physiol Gastrointest Liver Physiol 2006; 290: G232–G241.
Article CAS Google Scholar - Fortunato F, Kroemer G . Impaired autophagosome-lysosome fusion in the pathogenesis of pancreatitis. Autophagy 2009; 5: 850–853.
Article CAS Google Scholar - Kloppel G . Progression from acute to chronic pancreatitis. A pathologist's view. Surg Clin North Am 1999; 79: 801–814.
Article CAS Google Scholar - Lankisch PG, Brinkmann G, Maisonneuve P, Lowenfels AB . The natural history of acute pancreatitis: a long-term population-based study. Pancreas 2007; 35: 410.
Google Scholar - Nojgaard C, Becker U, Matzen P, Andersen JR, Holst C, Bendtsen F . Progression from acute to chronic pancreatitis: prognostic factors, mortality, and natural course. Pancreas 2011; 40: 1195–1200.
Article Google Scholar - Reszetow J, Hac S, Dobrowolski S, Stefaniak T, Wajda Z, Gruca Z et al. Biliary versus alcohol-related infected pancreatic necrosis: similarities and differences in the follow-up. Pancreas 2007; 35: 267–272.
Article Google Scholar - Bode C, Kugler V, Bode JC . Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 1987; 4: 8–14.
Article CAS Google Scholar - Bode JC, Bode C . Alkohol, Gastrointestinaltrakt und Pankreas. [Alcohol, the gastrointestinal tract and pancreas]. Ther Umsch 2000; 57: 212–219.
Article CAS Google Scholar - Boroni Moreira AP, de Cassia Goncalves Alfenas R . The influence of endotoxemia on the molecular mechanisms of insulin resistance. Nutr Hosp 2012; 27: 382–390.
CAS PubMed Google Scholar - Kelly CJ, Colgan SP, Frank DN . Of microbes and meals: the health consequences of dietary endotoxemia. Nutr Clin Pract 2012; 27: 215–225.
Article Google Scholar - Lieber CS, DeCarli LM . The feeding of ethanol in liquid diets. Alcohol Clin Exp Res 1986; 10: 550–553.
Article CAS Google Scholar - Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol 2011; 31: 379–446.
Article CAS Google Scholar - Kloppel G, Detlefsen S, Feyerabend B . Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch 2004; 445: 1–8.
Article Google Scholar - Fortunato F, Berger I, Gross ML, Rieger P, Buechler MW, Werner J . Immune-compromised state in the rat pancreas after chronic alcohol exposure: the role of peroxisome proliferator-activated receptor gamma. J Pathol 2007; 213: 441–452.
Article CAS Google Scholar - Tamizhselvi R, Shrivastava P, Koh YH, Zhang H, Bhatia M . Preprotachykinin-A gene deletion regulates hydrogen sulfide-induced toll-like receptor 4 signaling pathway in cerulein-treated pancreatic acinar cells. Pancreas 2011; 40: 444–452.
Article CAS Google Scholar - Zhou XY, Zhou ZG, Ding JL, Wang L, Wang R, Zhou B et al. TRAF6 as the key adaptor of TLR4 signaling pathway is involved in acute pancreatitis. Pancreas 2010; 39: 359–366.
Article CAS Google Scholar - Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E et al. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut 2009; 58: 813–819.
Article CAS Google Scholar - Ding SQ, Li Y, Zhou ZG, Wang C, Zhan L, Zhou B . Toll-like receptor 4-mediated apoptosis of pancreatic cells in cerulein-induced acute pancreatitis in mice. Hepatobiliary Pancreat Dis Int 2010; 9: 645–650.
CAS PubMed Google Scholar - Awla D, Abdulla A, Regner S, Thorlacius H . TLR4 but not TLR2 regulates inflammation and tissue damage in acute pancreatitis induced by retrograde infusion of taurocholate. Inflamm Res 2011; 60: 1093–1098.
Article CAS Google Scholar - Perejaslov A, Chooklin S, Bihalskyy I . Implication of interleukin 18 and intercellular adhesion molecule (ICAM)-1 in acute pancreatitis. Hepatogastroenterology 2008; 55: 1806–1813.
CAS PubMed Google Scholar - Ueda T, Takeyama Y, Yasuda T, Matsumura N, Sawa H, Nakajima T et al. Significant elevation of serum interleukin-18 levels in patients with acute pancreatitis. J Gastroenterol 2006; 41: 158–165.
Article CAS Google Scholar - Yuan BS, Zhu RM, Braddock M, Zhang XH, Shi W, Zheng MH . Interleukin-18: a pro-inflammatory cytokine that plays an important role in acute pancreatitis. Exp Opin Ther Targets 2007; 11: 1261–1271.
Article CAS Google Scholar - Paszkowski AS, Rau B, Mayer JM, Moller P, Beger HG . Therapeutic application of caspase 1/interleukin-1beta-converting enzyme inhibitor decreases the death rate in severe acute experimental pancreatitis. Ann Surg 2002; 235: 68–76.
Article Google Scholar - Grady T, Liang P, Ernst SA, Logsdon CD . Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 1997; 113: 1966–1975.
Article CAS Google Scholar - Sun J, Bhatia M . Blockade of neurokinin-1 receptor attenuates CC and CXC chemokine production in experimental acute pancreatitis and associated lung injury. Am J Physiol Gastrointest Liver Physiol 2007; 292: G143–G153.
Article CAS Google Scholar - Rongione AJ, Kusske AM, Kwan K, Ashley SW, Reber HA, McFadden DW . Interleukin 10 reduces the severity of acute pancreatitis in rats. Gastroenterology 1997; 112: 960–967.
Article CAS Google Scholar - Graf R, Schiesser M, Lussi A, Went P, Scheele GA, Bimmler D . Coordinate regulation of secretory stress proteins (PSP/reg, PAP I, PAP II, and PAP III) in the rat exocrine pancreas during experimental acute pancreatitis. J Surg Res 2002; 105: 136–144.
Article CAS Google Scholar - Schneider L, Hartwig W, Flemming T, Hackert T, Fortunato F, Heck M et al. Protective effects and anti-inflammatory pathways of exogenous calcitonin gene-related peptide in severe necrotizing pancreatitis. Pancreatology 2009; 9: 662–669.
Article CAS Google Scholar - Reding T, Bimmler D, Perren A, Sun LK, Fortunato F, Storni F et al. A selective COX-2 inhibitor suppresses chronic pancreatitis in an animal model (WBN/Kob rats): significant reduction of macrophage infiltration and fibrosis. Gut 2006; 55: 1165–1173.
Article CAS Google Scholar - Tartakoff AM, Jamieson JD . Subcellular fractionation of the pancreas. Methods Enzymol 1974; 31 (Pt A): 41–59.
Article CAS Google Scholar
Acknowledgements
This study was supported by a DFG Excellent Initiative Frontier award (FF) and institutional funding (MWB and JW). We would like to express our gratitude to Drs. Daniel Bimmler, Ion Deaciuc, Nathalia Giese, Rolf Graf and David J Hackam for their help and to Mrs. Karin Ruf, Zlata Antoni, Peter Rieger and Marta Bain for technical assistance. We are also indebted to the Pathophysiology Department at the University of Heidelberg for the IF microscopy support.
Author information
Authors and Affiliations
- Department of General, Visceral and Transplantation Surgery, Heidelberg, Germany
H Gu, J Werner, M W Büchler & F Fortunato - Institute of Pathology, University Clinic, Heidelberg, Germany
F Bergmann - Department of Gastroenterology, University of Pittsburgh, Pittsburgh, PA, USA
D C Whitcomb
Authors
- H Gu
- J Werner
- F Bergmann
- D C Whitcomb
- M W Büchler
- F Fortunato
Corresponding author
Correspondence toF Fortunato.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by Y Shi
Authors Contributions
Study concept, analysis and statistic: HG & FF Pathology: FB Supervision and funding: MWB, DCW, JW, and FF Manuscript writing: MWB, DCW, JW and FF This study was supported by a DFG Excellent Initiative Frontier award (FF) and institutional funding (MWB, DCW and JW).
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gu, H., Werner, J., Bergmann, F. et al. Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis.Cell Death Dis 4, e816 (2013). https://doi.org/10.1038/cddis.2013.354
- Received: 21 May 2013
- Revised: 15 August 2013
- Accepted: 26 August 2013
- Published: 03 October 2013
- Issue date: October 2013
- DOI: https://doi.org/10.1038/cddis.2013.354