Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger (original) (raw)
We report two NMR complex structures of PHDUHRF1 binding to unmodified or K9 trimethylated histone tails, which clarify a controversy regarding how the binding of UHRF1 to H3 tails is mediated. Based on our structures, H3R2, not H3K9, mediates PHD binding.
Human UHRF1, also known as ICBP90, plays central roles in maintaining CpG DNA methylation 1, 2 and epigenetic code inheritance 3. It harbors five recognizable functional domains: an ubiquitin-like domain (UBL) at the N-terminus, a tandem tudor domain (TudorUHRF1), a plant homeodomain (PHDUHRF1), a SET and RING associated (SRA) domain and a RING domain at the C-terminus, which mediate interactions of UHRF1 with histones and DNA (Figure 1A). Previous studies suggested that PHDUHRF1 defines binding specificity of UHRF1 to trimethylated H3 tail H3K9me3 3. However, recent studies showed that H3K9me3 binding is mediated by the TudorUHRF1 4, 5. PHDUHRF1 is structurally different from the canonical PHD fingers (Figure 1). It has additional Cys residues at its amino terminal portion; it is also devoid of the residues to form the H3K4me3-binding aromatic cage 6, 7, 8, 9 and a terminal aspartic acid residue to recognize H3K4me0 10, 11, 12, 13. Therefore, exactly how PHDUHRF1 binds to the N-terminal H3 tail remains unclear.
Figure 1
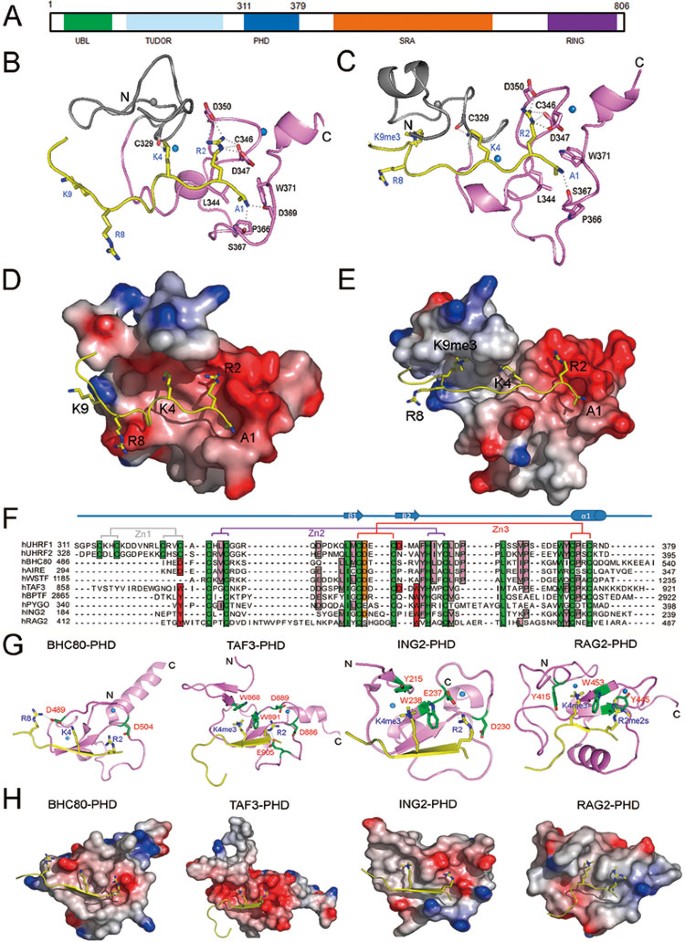
(A) Schematic representation of the functional domains in the human UHRF1 protein. (B, C) Ribbon representation of the complexes PHDUHRF1-H3K9me0 and PHDUHRF1-H3K9me3 highlights the secondary structural elements (protein, violet; peptide, yellow). The hydrogen bonds are displayed as dashed lines. For clarity, the first zinc finger is displayed in grey and spheres are zinc ions. (D, E) Electrostatic potential surface of the complexes PHDUHRF1-H3K9me0 and PHDUHRF1-H3K9me3, where PHDUHRF1 is shown as surface representation and H3 peptide, is shown as ribbon representation and colored in yellow. Residues H3R2, H3K4, H3R8 and H3K9 are indicated as stick representation. (F) Sequence alignment of PHD from representative PHD-containing proteins. Zn-chelating residues and highly conserved residues are highlighted with dark green background, the characteristic histone peptide interaction positions are in red background, the residues Asp that usually interacts with H3R2 are in brown background and conserved residues are in pink background. Secondary structural elements are indicated above the sequences. Residues that involved in zinc coordination are connected by solid lines (the first, second and third tetrads are connected by gray, purple and red lines, respectively). (G, H) Structure comparison of PHDUHRF1-H3 complex with other PHD-H3 complexes: PHDBHC80-H3 (2PUY), PHDTAF3-H3K4me3 (2K17), PHDING2-H3K4me3 (2G6Q) and PHDRAG2-H3R2me2aK4me2 (2V87). All histone peptides are colored in yellow. Zinc atoms are indicated as balls. The side chains of residues for specific recognition are shown as stick representation (green and labeled with red characters) and hydrogen bonds are shown as dashed lines.
To address this issue, we determined 3D NMR structures of free PHDUHRF1, and two complexes, namely PHDUHRF1-H3K9me0 and PHDUHRF1-H3K9me3 (Figure 1 and Supplementary information, Data S1, Figure S1, Table S2). In these three structures, PHDUHRF1 is coordinated by three zinc ions to form a rod-shape structure, containing a small α-helix, a double-stranded anti-parallel β-sheet and three loops. The free and bound PHDUHRF1 structures have backbone atoms RMSD value of 1.7 Å, indicating that H3 peptides binding does not induce overall major conformational changes of PHDUHRF1. The C-terminus of PHDUHRF1 is coordinated by two zinc ions in an interleaved, compact manner, which is similar to the reported histone tail-binding PHD domain structures (Figure 1) 6, 7, 8, 9, 10, 11, 12, 13. However, a difference lies at the N-terminus of the PHDUHRF1, where the first zinc ion coordinates the N-terminal loop region. Such a structural feature is not found in other histone-binding PHD domains 6, 7, 8, 9, 10, 11, 12, 13, although the function of this loop remains unknown.
In both structures of PHDUHRF1-H3K9me0 and PHDUHRF1-H3K9me3 (Figure 1B-1E), the N terminus of H3 peptide is anchored through nonpolar interactions between methyl group of H3A1 and hydrophobic side chains of PHDUHRF1 residues L344, P366 and W371 and hydrogen bonds between H3A1 nitrogen atom and backbone oxygen of PHDUHRF1 P366 and D369. Apparently, H3 residues R8 and K9 (unmethylated or trimethylated) have no interaction with PHDUHRF1, with their side-chains extending outwards. Recently, it was reported that H3K9me3 is recognized via tandem Tudor domain 4, 5, this may explain why the side chain of H3K9 does not make contact with PHDUHRF1 in our complex structures. The first zinc finger almost has no contact with histone peptides (Figure 1B-1E), except that backbone oxygen atom of PHDUHRF1 C329 forms a hydrogen bond with the side-chain nitrogen atom (Nξ) of H3K4. The second zinc finger seems to stabilize the overall structure only. Both H3K9me0 and H3k9me3 peptides are stapled to the protein surface with intermolecular hydrogen bonds network mainly formed by H3 peptides H3R2 residue and protein residues C346, D347 and D350 in zinc finger 3 of PHDUHRF1. The R2 side-chain of H3 is docked into the negatively charged groove in zinc finger 3 of PHDUHRF1 by forming hydrogen bonds network with the oxygen atoms of the side chains of D347 and D350, and probably the backbone oxygen of C346, which supports H3R2 acting as a major contact site. Collectively, these results indicate that H3R2 residue plays a major role in PHDUHRF1-mediated histone H3 recognition.
Although PHDUHRF1 shows similar overall structures to those of other histone tail-binding PHD domains (Figure 1G-1H), the PHD domains of BPTF 6, 7, YNG1 13 and ING2 8, 9 that recognize trimethylated H3K4 are characterized by an aromatic cage, while binding of unmethylated K4 of the H3 tail by the PHD domains of BHC80 12 and AIRE 10, 11 is mediated by intermolecular hydrogen bonds using a terminal aspartic acid residue. Therefore, in our studies, the PHDUHRF1 R2 recognition mediated predominantly by hydrogen bonds represents a new model of H3 tail recognition.
To confirm the binding model revealed by the structures above, we carried out NMR titration binding assay and in vitro fluorescence binding assay (Supplementary information, Table S1, Figures S2 and S3). On binding to H3K9me0 and H3K9me3, the chemical shifts of backbone HN and N atoms of PHDUHRF1 are changed in a similar manner (Supplementary information, Figure S4). Both H3 peptides do not arouse obvious variance of the chemical shift of residues in the first zinc finger, indicating that the first zinc finger is not involved in the interaction. The cross peak of D350 disappears in 1H-15N HSQC spectra of PHDUHRF1-H3K9me0 and PHDUHRF1-H3K9me3, even when the molar ratio of PHDUHRF1:H3K9me0 or H3K9me3 is set as 1:0.2, which implies that rigid hydrogen bond net may occur between PHDUHRF1 D350 side-chain and H3R2 positively charged side chain (PHDUHRF1 and PHDUHRF1 D350A mutant have binding affinities to unmodified H3 peptide, at ∼4 μM and ∼17 μM, respectively.), as predicted in the structures. Their 2D 1H-15N HSQC spectra, however, superimpose well, except for minor differences in the interaction sites including D347, C346 and C329, but both spectra are in sharp contrast to that of free PHDUHRF1 (Supplementary information, Figure S3A), suggesting that K9 trimethylation has little effect on binding of H3 tail to PHDUHRF1 (both H3K9me0 and H3K9me3 have _K_D values of ∼4 μM to PHDUHRF1). This observation is consistent with the fact that ∼73% of H3K9me3 and ∼70% of H3k9me0 solvent-accessible surfaces are buried upon binding to PHDUHRF1. Most 1H-15N HSQC cross peaks of PHDUHRF1-H3R2me2s and PHDUHRF1-H3R2me2a do not overlap well with those of PHDUHRF1-unmodified H3 peptide (Supplementary information, Figure S3B and S3C), but they are still a little different from those of free PHDUHRF1 (Supplementary information, Figure S3E), indicating that dimethylation of H3R2 disrupts to large extent the interaction between PHDUHRf1 and H3 peptides. Especially, upon binding to symmetrically or asymmetrically dimethylated H3R2, PHDUHRF1 D347 cross peak moves to the position close to that of D347 in free PHDUHRF1 (Supplementary information, Figure S2C). In contrast, monomethylation at H3R2 just results in milder effect on binding, as D347 cross peak of PHDUHRF1-H3R2me1 is still quite close to that of PHDUHRF1-H3 (Supplementary information, Figure S3D). These data are consistent with the following _K_D measurements: (1) mutations from D347 to A347 (i.e. D347A) in PHDUHRF1, as well as from R2 to A2 (i.e. R2A) in H3 tail cause a binding affinity that is too weak to be measured; (2) both symmetric and asymmetric dimethylation at R2 have a significant impact on binding (_K_D reduction of 10-14-fold, Supplementary information, Figure S3C and Table S1), while monomethylation at H3R2 has a milder effect (_K_D reduction of ∼8-fold, Supplementary information, Table S1). We thus conclude that H3R2 interacts with PHDUHRF1 D347 residue, but dimethylation of H3R2 residue impedes this interaction. We then tested the effects on the binding by methylation at H3K4. Interestingly, obvious position shift of C329 cross peak is found in case of PHDUHRF1-H3K4me2, but not observed in PHDUHRF1 with any other H3 peptides, suggesting that H3K4 residue may interact with PHDUHRF1 C329 during the interaction, consistent with the findings from 3D structures. In 2D 1H-15N HSQC spectrum of PHDUHRF1-H3K4me2, the positions of cross peaks of C346, D347 and D350 do not change much compared to those of PHDUHRF1-unmodified H3 (Supplementary information, Figure S3F), indicative of no significant changes in binding caused by H3K4 dimethylation (_K_D values, PHDUHRF1-H3K4me2: ∼10 μM and PHDUHRF1-H3K4me1: ∼7 μM, respectively). But trimethylation at H3K4 decreases the binding affinity by ∼6-fold (_K_D value ∼26 μM), which might be due to clash of the third methyl group of H3K4me3 with the side-chains of residues L328 and R330, or C329 backbone oxygen, as discussed in PHDBHC80-H3K4 complex structure 12. Taken together, these data demonstrate that dimethylation at H3R2 significantly influences binding of H3 tail to PHDUHRF1, while monomethylation at H3R2 and methylation at H3K4 have a slight impact, and trimethylation at H3K9 does not affect the binding. Therefore, H3R2 may be a critical contact site for UHRF1. In other words, these results suggest that PHDUHRF1 binds to histone tail H3 via recognition of the unmodified R2 residue.
Since TudorUHRF1 was reported to specifically recognize K9me3 site of the H3 tail 4, 5, we further investigated the binding affinities of full-length UHRF1 and (Tudor-PHD)UHRF1 to unmodified or K9-trimethylated H3 peptides (Supplementary information, Table S1). The _K_D values of (Tudor-PHD)UHRF1 binding to unmodified or modified H3 tail are ∼4 μM and ∼10 μM, respectively, comparable to the value (∼4 μM) of PHDUHRF1 binding to H3 N-tail; while full-length UHRF1 has binding affinities of ∼3 μM to K9-trimethylated H3 tail, a little stronger than that (∼5 μM) to unmodified H3 peptide. We speculate that in full-length UHRF1, PHDUHRF1 and TudorUHRF1 might interact with H3K9me3 peptide better than in (Tudor-PHD)UHRF1. The detailed mechanism of whether and how these two domains within UHRF1 accommodate one H3 peptide need to be further probed, as well as the functional roles of reading of unmodified H3R2 by PHDUHRF1 in vivo.