Generation of gene-modified mice via Cas9/RNA-mediated gene targeting (original) (raw)
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) adaptive immune systems are found in bacteria and archaea to protect the hosts against the invasion of viruses and plasmids1,2,3. Three types (I-III) of CRISPR systems with different features have been identified. The CRISPR-associated protein Cas9 that belongs to the type II CRISPR/Cas system has attracted much attention due to its potential use in genomic engineering. Cas9 contains one HNH motif and three RuvC-like motifs, homologous to HNH and RuvC endonucleases, respectively (Supplementary information, Figure S1)4,5,6,7,8. Recent studies showed that Cas9 displayed strong DNA cleavage activity in bacteria and in test tubes. Its nuclease activity is guided by two non-coding RNA elements of the system; one is crRNA (CRISPR RNA) that contains about 20 base pairs (bp) of unique target sequence (called spacer sequence) and the other is tracrRNA (trans-activating crRNA). These two RNA elements form a crRNA:tracrRNA duplex that directs Cas9 to target DNA via complementary base pairing between the spacer on the crRNA and the complementary sequence (called protospacer) on the target DNA. The 3 nucleotides (nt) located immediately at the 3′ side next to the protospacer sequence constitute the protospacer adjacent motif (PAM) that is required to ensure the cleavage specificity in target sequences9,10.
Theoretically, CRISPR systems can be used in higher eukaryotes through ectopically expressing the enzyme (Cas9) and the RNAs, much like the zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) (Figure 1A). To test this, we first asked whether Cas9 could perform DNA cleavage in zebrafish. We generated codon-optimized Cas9 (Supplementary information, Table S1 and Data S2) and cloned it into an expression vector. A chimeric RNA that is a single engineered RNA molecule combining features of both crRNA and tracrRNA9 was designed to target one of the two sites (EGFP-A and -B) in the pEGFP-N1 plasmid (Figure 1B, Supplementary information, Figures S2 and S3). DNA fragments corresponding to Cas9 and the chimeric RNA were transcribed to RNAs in vitro by the T7 RNA polymerase and the Cas9 RNA was further modified by adding a translation-required cap at the 5′-end and a poly-A tail at the 3′-end to make it resemble an authentic mRNA. The Cas9 mRNA, chimeric RNA, and pEGFP-N1 plasmid were co-injected into one-cell zebrafish embryos. Similar strategies have previously been employed to test ZFN and TALEN activity11,12. The embryos were cultured for 12 h before being harvested for total DNA extraction.
Figure 1
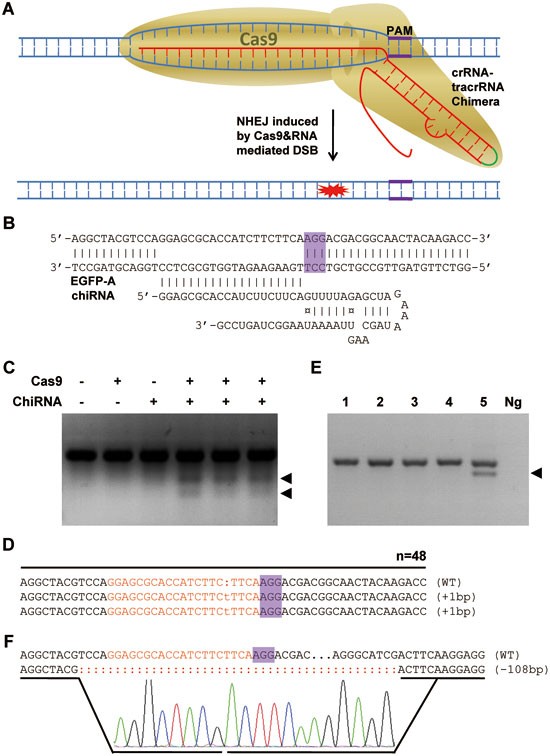
Cas9/RNA-mediated gene targeting. (A) Schematic diagram of Cas9/RNA-mediated gene targeting. Chimeric RNA, a single engineered RNA molecule combining crRNA and tracrRNA, can guide Cas9 to cleave the target site of ∼20 nt. PAM, an NGG motif shown in purple, is essential for the activity of the complex. (B) Schematic representation of EGFP-A chimeric RNA (chiRNA) binding to pEGFP-N1 plasmid through spacer sequence. PAM is highlighted in purple. (C) Chimeric RNA guides Cas9 to cleave pEGFP-N1 at the target site in zebrafish. Four hundred nanograms per litre of Cas9 mRNA, 100 ng/μl of EGFP-A chimeric RNA, and pEGFP-N1 plasmid were co-injected into one-cell zebrafish embroys. Cleavage assays were performed 12 h after injection. Arrowheads indicate that two cleavage bands (about 295 bp and 198 bp) were detected in zygotes treated with Cas9 and non-annealed EGFP-A chimeric RNA. The Cas9 and chimeric RNA were added as indicated. (D) Sequencing results of T-A colonies of targeted fragments amplified from the sample of lane 4 in C. Two colonies with 1-bp insertion were detected in 48 colonies. Target sequence is capitalized and highlighted in red. Red lower case represents insert sequence. (E) PCR amplification of targeted fragment in the EGFP gene in founder mice treated with Cas9/RNA microinjection. Founder mice were generated as described in Supplementary information, Data S1. PCR amplification of the targeted fragment was performed using genomic DNA extracted from the tails of the founders as templates. Primers used were listed in Supplementary information, Figure S2. Arrowhead indicates a truncated band in founder #5. Ng, negative control. (F) The PCR products from founder #5 were subjected to T-A cloning. Twenty colonies were randomly selected for DNA sequencing. A 108-bp deletion was detected in nine colonies. Target sequence is capitalized and highlighted in red.
The targeted region on the EGFP plasmid was amplified from the extracted DNA; the purified PCR products were then denatured and reannealed to form hybridized DNA, followed by digestion with the T7 endonuclease 1 (T7EN1)13 that can recognize and cleave mismatched DNA (Supplementary information, Figure S4). Gel electrophoresis of T7EN1-digestion products clearly showed two smaller fragments besides the amplicon of the targeted region, which were not observed in the control (Figure 1C and Supplementary information, Figure S3), suggesting that the target DNA was cleaved by Cas9. To precisely locate the site of cleavage, we cloned the PCR products and analyzed the clones by DNA sequencing. There were 2 mutant clones out of 48 sequenced. The cutting occurred about 4 nt away from the PAM (Figure 1D). These results demonstrate that Cas9/RNA can site-specifically cut DNA in eukaryotic cells.
To improve the cutting efficiency of the Cas9 and chimeric RNA module, we annealed the chimeric RNA before injection to facilitate its correct folding. Indeed, T7EN1 cleavage assay showed that using preannealed chimeric RNA in the Cas9 system generated stronger cleavage bands. Consistently, more mutants (3 out of 46 PCR clones) were detected by DNA sequencing. Similarly, the cutting occurred about 4 nt away from the PAM (Supplementary information, Figure S5). These results suggest that the conformation of the chimeric RNA probably affects the activity of the Cas9 complex.
Different from prokaryotes, targeting eukaryotic DNA requires nuclear translocation of the nucleases. As expected, when expressed in the mammalian cell line 293T, the prokaryote-derived Cas9 was only detected in cytoplasm despite the addition of an SV40 nuclear localization signal (NLS) to the N- or both the N- and C-termini of Cas9. Even the addition of a triple NLS to the N-terminus did not work (Supplementary information, Figure S6). The reason might be that the NLS peptide was buried or shielded during the folding of the Cas9 protein. To circumvent this, we added a linker (32 amino acids) between the NLS and Cas9, which resulted in the successful nuclear localization of Cas9 (Supplementary information, Figure S6). We tested the cleavage activity of all tagged-Cas9s and found that, as expected, NLS-flag-linker-Cas9 displayed enhanced cleavage activity (Supplementary information, Figure S7).
The cleavage of exogenous DNA in zebrafish embryos encouraged us to test whether Cas9/RNA could be used to site-specifically disrupt endogenous genes in mice, as has been achieved with ZFN and TALEN. We first used the Pouf5-IRES-EGFP knock-in mouse line that carries one copy of the EGFP gene incorporated into the mouse genome. Twenty nanograms per microlitre of NLS-flag-linker-Cas9 mRNA and 20 ng/μl preannealed EGFP-A chimeric RNA were co-injected into one-cell mouse embryos obtained from the crosses between male homozygous Pouf5-IRES-EGFP knock-in mice and superovulated C57BL/6J female mice. Twenty-four injected embryos were transferred into pseudopregnant CD1 female mice and five live animals were born so far. As the Pouf5-IRES-EGFP male mice that we used were homozygous for the knock-in gene, all five founder mice were, as expected, genotyped as heterozygous, indicated by PCR amplification of a knock-in fragment using genomic DNA extracted from the tails of the founders 5 days after birth (Supplementary information, Figure S8). Site-specific cleavage of the endogenous EGFP locus was first analyzed by PCR amplification of the target site using the same tail genomic DNA as used in genotyping. As shown in Figure 1E, a smaller amplicon appeared in founder #5, whereas no mutant bands were detected in the PCR products of founders #1-4. Twenty T-A colonies of the PCR products from founder #5 were randomly picked for DNA sequencing. A 108-bp deletion was detected in nine colonies (Figure 1F). No mutations were detected in the remaining 11 colonies (data not shown). Further analyses by the T7EN1 cleavage assay and DNA sequencing did not detect mutations of the target region in founders #1-4 (data not shown). These results indicate that the Cas9/RNA system induced a site-specific cleavage on endogenous single-copied EGFP. Given the presence of the wild-type amplicon besides the mutant one, fonder #5 is mosaic for the mutant EGFP, which suggests that the cleavage and subsequent repair occurred after the first division of the injected one-cell embryo. It is noteworthy that similar to ZFN and TALEN, Cas9/RNA-induced double-stranded break leads to nonhomologous end joining-mediated repair, which generates indels (insertions or deletions) of different sizes around the DNA break site. The 108-bp deletion within the EGFP gene in the mouse model only represents one case; it is likely that more indels of different sizes would be found upon screenings of more founder mice treated with Cas9/RNA.
Next, we tested the efficiency of the same targeting strategy in another EGFP mouse line (CAG-EGFP, with multiple copies of the transgene). Seven pups were born from 51 injected embryos and one of them (#4) contained disrupted EGFP as shown by the T7EN1 cleavage assay (Supplementary information, Figure S9). PCR products from #4 were cloned and sequenced. Eleven out of 50 clones contained mutations. Interestingly, we detected six mutant forms, indicating that Cas9/RNA-mediated cleavage had occurred multiple times on different copies of the EGFP transgene in the same embryo (Supplementary information, Figure S9). Taken together, we demonstrated that Cas9/RNA can induce site-specific cleavage of chromosomal loci in the mouse genome.
In summary, our findings demonstrate that Cas9/RNA can site-specifically cut DNA in zebrafish and mouse embryos, paving the way for its use in the generation of gene-disrupted animals. During the preparation of this manuscript, two papers were published in Science, reporting the use of the Cas9/RNA system in mammalian cells14,15. Our study goes beyond cultured cells and shows the utility of the Cas9/RNA as a genetic tool in generating genetically engineered mice and, potentially, other mammals. Different from ZFN and TALEN, the Cas9/RNA system is RNA-guided and easy to engineer, offering better flexibility as a genomic engineering tool. The fact that this system uses RNA to guide DNA cleavage points to its potential use in large-scale genetic screenings, as a library of guide RNAs can be constructed.