Intellectual disability without epilepsy associated with STXBP1 disruption (original) (raw)
Abstract
STXBP1 (Munc18-1) is a component of the machinery involved in the fusion of secretory vesicles to the presynaptic membrane for the release of neurotransmitters. De novo missense mutations in STXBP1 were recently reported in patients with Ohtahara syndrome, a form of encephalopathy with severe early-onset epilepsy. In addition, sequencing of the coding region of STXBP1 in 95 patients with non-syndromic intellectual disability (NSID) revealed de novo truncating mutations in two patients who also showed severe non-specific epilepsy, suggesting that STXBP1 disruption has the potential of causing a wide spectrum of epileptic disorders in association with intellectual disability. Here, we report on the mutational screening of STXBP1 in a different series of 50 patients with NSID and the identification of a novel de novo truncating mutation (c.1206delT/ p.Y402X) in a male with NSID, but surprisingly with no history of epilepsy. This is the first report of a patient with a truncating mutation in STXBP1 that does not show epilepsy, thus, expanding the clinical spectrum associated with STXBP1 disruption.
Similar content being viewed by others
Introduction
A growing body of work suggests that disruption of synapse development or function explains a large fraction of patients with intellectual disability (ID; also referred as mental retardation).1, 2 One of the key events of synaptic transmission is the fusion of secretory vesicles to the presynaptic membrane leading to the release of the neurotransmitters into the synaptic cleft. This process is mediated by a complex composed of SNARE proteins and of Munc18-1/STXBP1.3, 4, 5 Consistent with its important role for neurotransmission, recent studies showed that mutations in STXBP1 cause severe ID and epilepsy.6, 7 Using array genomic hybridization, Saitsu et al6 identified a de novo 2 Mb deletion encompassing STXBP1 in a patient with Ohtahara syndrome, an infantile form of epileptic encephalopathy characterized by burst suppressions and severe ID. Analysis of candidate genes mapped to the deletion revealed missense mutations in STXBP1 in four other patients with Ohtahara syndrome. These mutations were found to arise de novo in at least three of these cases and to disrupt STXBP1 biochemical properties.6 Subsequently, our group reported on the identification of de novo truncating mutations in STXBP1 in two patients with severe non-syndromic ID (NSID) and intractable partial complex epilepsy.7 In order to better understand the contribution of STXBP1 mutations to NSID, we studied 50 additional patients and identified a novel de novo STXBP1 truncating mutation in a patient with NSID, but surprisingly with no history of epilepsy. This observation indicates that STXBP1 mutations are associated with a wide spectrum of phenotypes, from NSID with or without epilepsy to syndromic forms of epilepsy.
Methods
NSID subjects
We recruited 50 sporadic cases (24 males and 26 females) with unexplained NSID, the majority of which were of French Canadian origin. We selected sporadic cases to increase the likelihood of finding de novo mutation. In the context of this study, and of our previous one,7 NSID was defined using the following criteria: (1) diagnosis of ID established on a clinical basis using standardized developmental or IQ tests; (2) absence of specific dysmorphic features, as assessed by an experienced clinical geneticist; (3) birth weight and postnatal growth within normal limits; (4) normal head circumference at birth; (5) absence of risk factors such as neonatal asphyxia, prematurity or exposure to teratogenic drugs and (6) negative standard investigations, including comparative genomic hybridization studies, molecular testing for the common expansion mutation in FMR1 and brain CT-scan or MRI. A subset of these NSID patients (_n_=11) displayed epilepsy. Blood samples were collected from all members of the NSID cohort, as well as from their parents after approval by the Sainte Justine Hospital Research Center Medical Ethics Committee for genetic studies. Written consent forms were obtained from all guardians (parents) of the patients participating in this study.
STXBP1 sequencing
Genomic DNA was extracted from blood samples using the Puregene DNA kit (Gentra System; Qiagen, Mississauga, ON, Canada). Paternity and maternity of each NSID patient were confirmed using six informative unlinked microsatellite markers. PCR amplification and sequencing of all of the STXBP1 exons (19 in total) and intronic splice junctions based on the structure of the longest isoform (Refseq no. NM_001032221) were performed as previously described.7
Results
We sequenced all STXBP1 exons and splice junction boundaries from the genomic DNA of 50 patients with NSID. We identified a heterozygous 1-bp deletion that creates a premature stop codon at amino acid position 402 (c.1206delT/p.Y402X) in one of the patients. This mutation was not identified in the DNA of both parents, indicating that it originated de novo (Figure 1). It lies in domain-3 of STXBP1, in close proximity to a previously reported de novo truncating mutation (p.R388X) identified by our group in a patient with NSID and severe epilepsy (Figure 1).7 Domain-3 together with domain-1 of STXBP1 form the central cavity providing surfaces for Syntaxin-1 binding, an essential step for SNARE complex assembly and subsequent neurotransmitter release.3, 8 No other amino acid altering or splicing mutations were identified in STXBP1 in the remaining patients. We previously sequenced STXBP1 in healthy French Canadian controls (_n_=190) and did not identify any amino acid changing or splicing mutations.7
Figure 1
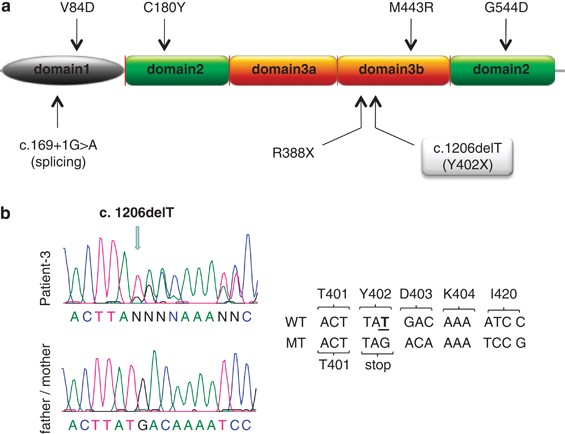
Pathogenic STXBP1 mutations. (a) Positions of the reported pathogenic STXBP1 mutations. Missense mutations reported to cause Ohatahra syndrome6 are indicated on top of the STXBP1 sequence, while splicing and truncating mutations reported in NSID with nonspecific epilepsy7 (c.169+1G>A and p.R388X) or without epilepsy (current study, p.Y402X, highlighted) are shown below the STXBP1 sequence. The positions of the various STXBP1 protein domains are depicted based on the rat STXBP1 crystal structure.8 (b) Chromatograms of the STXBP1 de novo mutation identified in patient 3 and the corresponding representative sequence from parents of patient 3 (both the mother's and father's STXBP1 sequences lack c.1206delT mutation). Wild-type (WT) and mutant (MT) STXBP1 DNA sequences are shown along with their corresponding amino acids. Numbering of the coding nucleotides and amino acids are based on isoform a of STXBP1 (Refseq NM_003165).
The patient with the c.1206delT (referred to as patient 3) is a man, aged 21 years, born to non-consanguineous French Canadian parents with no family history of developmental disabilities (Table 1). He was delivered at term after an unremarkable pregnancy. Immediate neonatal course was uneventful. His psychomotor development was characterized by global delay. He first walked at 2 years of age. He could only say 20 words at 4 years of age. Cognitive evaluation with the Leiter cognitive test was not possible at that age because he did not understand the tasks. Another attempt to evaluate his cognitive profile with the Leiter and the WPPSI-R tests also failed at 5 years and 9 months for the same reason. Currently, he can use short and incomplete sentences to communicate his needs. He can evoke events of the past, but he cannot have a conversation. He can dress by himself, use tools and play simple video games but he cannot draw, cut his meals or tie his shoes. He is treated with methylphenidate for attention disorder without hyperactivity or impulsivity. There is no history of seizures. Physical examination did not reveal any specific dysmorphic features. Neurological exam showed the presence of diffuse tremor of the extremities that was increased upon voluntary movements, likely contributing to his poor fine motor skills. His tonus is normal. There was no dysmetria or ataxia. He walks with an increased polygon of sustentation without retropulsions. Electroencephalography performed at 21 years of age did not reveal any abnormality except for intermittent slow dysfunction in the left temporal area. Brain CT scan performed at 4 years of age was normal.
Table 1 Clinical profiles of patients with de novo truncating STXBP1 mutations
Discussion
We report here, a novel de novo truncating mutation, c.1206delT/p.402X, in a NSID patient with severe cognitive deficit, but with no history of epilepsy. This mutation is likely to be pathogenic as it is predicted to truncate STXBP1 towards the middle of its sequence, in the same functional domain as p.R388X, which was recently reported in a patient with severe NSID and non-syndromic epilepsy.7 This conclusion is further supported by the observations that truncation of the Caenorhabditis elegans orthologue of STXBP1 downstream of position p.Y402 results in defects in synaptic vesicle docking9 and that Stxbp1 haploinsufficiency causes impaired neurotransmission in mice.10
All seven known patients with pathogenic point mutations in STXBP1 show severe ID. Four of them, all carrying missense mutations, presented with Ohtahara syndrome and, with one exception, developed infantile spasms,6 whereas two other patients, bearing truncating mutations, show severe NSID with non-syndromic intractable partial complex epilepsy,7 indicating that mutations in STXBP1 can result in different types of epileptic manifestations. The case described here shows that STXBP1 disruption can also cause severe ID without epilepsy. Additional clinical clues suggesting STXBP1 mutations include the presence of tremor, hypotonia and hyperventilation, which were observed in a subset of patients (Table 1).6, 7
Although the small number of patients identified at this point in time precludes drawing definitive correlations between genotypes and phenotypes, mutations leading to a decrease of STXBP1 function appear to be associated with variable expressivity. First, different genetic lesions in STXBP1 can result in the same phenotype. For instance, although missense mutations were exclusively identified in patients with Ohtahara syndrome, another patient with this condition was found to carry a de novo 2 Mb deletion encompassing STXBP1.6 Second, similar genetic lesions in STXBP1 can lead to different phenotypes, as illustrated by the variable presence of severe epilepsy in our patients with truncating mutations (Table 1).
By combining this study and our previous one, we have identified three de novo truncating STXBP1 mutations in 145 sporadic NSID cases (∼2%). This high rate of deleterious mutations suggests that analysis of STXBP1 should be considered in sporadic cases of severe NSID, with or without epilepsy.
References
- Chelly J, Khelfaoui M, Francis F, Cherif B, Bienvenu T : Genetics and pathophysiology of mental retardation. Eur J Hum Genet 2006; 14: 701–713.
Article CAS Google Scholar - Humeau Y, Gambino F, Chelly J, Vitale N : X-linked mental retardation: focus on synaptic function and plasticity. J Neurochem 2009; 109: 1–14.
Article CAS Google Scholar - Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D : Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. Embo J 2008; 27: 923–933.
Article CAS Google Scholar - Fisher RJ, Pevsner J, Burgoyne RD : Control of fusion pore dynamics during exocytosis by Munc18. Science 2001; 291: 875–878.
Article CAS Google Scholar - Gerber SH, Rah JC, Min SW et al: Conformational Switch of Syntaxin-1 Controls Synaptic Vesicle Fusion. Science 2008; 321: 1507–1510.
Article CAS Google Scholar - Saitsu H, Kato M, Mizuguchi T et al: De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40: 782–788.
Article CAS Google Scholar - Hamdan FF, Piton A, Gauthier J et al: De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009; 65: 748–753.
Article CAS Google Scholar - Misura KM, Scheller RH, Weis WI : Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 2000; 404: 355–362.
Article CAS Google Scholar - Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM : Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci 2003; 6: 1023–1030.
Article CAS Google Scholar - Toonen RF, Wierda K, Sons MS et al: Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci USA 2006; 103: 18332–18337.
Article CAS Google Scholar
Acknowledgements
We thank the patients and their parents for participating to this study. We are grateful for the dedicated work of members of the S2D team (CHUM Research Center, Montreal), including management (Claude Marineau and Ronald G Lafrenière), bioinformatics (Edouard Henrion, Ousmane Diallo and Dan Spiegelman) and genetic screening divisions (Amelie Piton, Annie Raymond, Annie Levert, Pascale Thibodeau, Sandra Laurent and Karine Lachapelle). We are also thankful for the McGill University and Génome Québec Innovation Centre Sequencing (Pierre Lepage, Sébastien Brunet and Hao Fan Yam) and Bioinformatics (Louis Létourneau and Louis Dumond Joseph) groups. Supported by grants from the Canadian Institute of Health Research (CIHR; to JLM, GAR and JCL), Réseau de Génétique Médicale Appliquée/Fonds de la Recherche en Santé (FRSQ; to JLM), Genome Canada and Genome Quebec and co-funding by Université de Montréal for the S2D project (to GAR). JLM is a recipient of a Clinical Investigatorship Award of the CIHR (Institute of Genetics) and of a Senior Scientist Award from the FRSQ.
Author information
Authors and Affiliations
- The Centre of Excellence in Neuromics of Université de Montréal and Synapse to Disease (S2D) group, Montréal, Quebec, Canada
Fadi F Hamdan, Julie Gauthier, Sylvia Dobrzeniecka, Laurent Mottron, Guy A Rouleau & Jacques L Michaud - Research Center CHU Sainte-Justine, Montréal, Quebec, Canada
Fadi F Hamdan, Anne Lortie, Michel Vanasse, Guy D'Anjou & Jacques L Michaud - CHUM Research Center and Department of Medicine, Université de Montréal, Montreal, Canada
Julie Gauthier, Sylvia Dobrzeniecka & Guy A Rouleau - Centre de Recherche Fernand-Séguin, Hôpital Rivière-des-Prairies, Montréal, Quebec, Canada
Laurent Mottron - Department of Physiology, Le Groupe de Recherche sur le Système Nerveux Central, Université de Montréal, Montreal, Quebec, Canada
Jean Claude Lacaille
Authors
- Fadi F Hamdan
You can also search for this author inPubMed Google Scholar - Julie Gauthier
You can also search for this author inPubMed Google Scholar - Sylvia Dobrzeniecka
You can also search for this author inPubMed Google Scholar - Anne Lortie
You can also search for this author inPubMed Google Scholar - Laurent Mottron
You can also search for this author inPubMed Google Scholar - Michel Vanasse
You can also search for this author inPubMed Google Scholar - Guy D'Anjou
You can also search for this author inPubMed Google Scholar - Jean Claude Lacaille
You can also search for this author inPubMed Google Scholar - Guy A Rouleau
You can also search for this author inPubMed Google Scholar - Jacques L Michaud
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJacques L Michaud.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hamdan, F., Gauthier, J., Dobrzeniecka, S. et al. Intellectual disability without epilepsy associated with STXBP1 disruption.Eur J Hum Genet 19, 607–609 (2011). https://doi.org/10.1038/ejhg.2010.183
- Received: 01 June 2010
- Revised: 06 September 2010
- Accepted: 07 October 2010
- Published: 02 March 2011
- Issue Date: May 2011
- DOI: https://doi.org/10.1038/ejhg.2010.183