Mitochondria in innate immunity (original) (raw)
Mitochondria control host cell metabolic processes and apoptosis. In addition, they are emerging as crucial hubs for innate immune signalling. Several key innate immune mediators are functionally associated with these organelles, and mitochondrial alarmins can trigger inflammation.
Introduction
Innate immune responses in mammalian cells rely on the detection of conserved molecular motifs found in microbial pathogens and known as ‘pathogen‐associated molecular patterns (PAMPs)’, which include viral nucleic acids as well as bacterial molecules such as lipopolysaccharide, lipoproteins, flagellin, peptidoglycan and CpG DNA (Medzhitov, 2007). In the past few years, it has also become clear that non‐microbial danger signals—composed of host molecules often released by necrotic cells in the context of tissue damage and known as ‘danger‐associated molecular patterns (DAMPs)’—also trigger innate immune signalling and promote inflammation (Benko et al, 2008). Both PAMPs and DAMPs trigger innate immunity following detection by several families of host sensor molecules, collectively called pattern recognition receptors (PRRs). The most‐studied families of mammalian PRRs include Toll‐like receptors (TLRs; Akira & Takeda, 2004), Nod‐like receptors (NLRs; Fritz et al, 2006) and RIGI‐like receptors (RLRs; Takeuchi & Akira, 2009). TLRs are membrane‐spanning molecules that detect extracellular or luminal PAMPs and DAMPs, whereas NLRs and RLRs sense intracellular molecular signatures. Once activated, PRRs trigger specific host defence signalling, the most commonly studied of which are pro‐inflammatory pathways that activate nuclear factor κB (NF‐κB), in response to most TLRs, the NLR family members NOD1 and NOD2, and RLRs; caspase 1 inflammasome, activated by the NLRP subfamily of NLR proteins and RLRs; and type I interferon (Elinav et al, 2011; Kawai & Akira, 2011; Loo & Gale, 2011). Finally, several new PRRs have been identified recently that detect intracellular foreign DNA, and these include AIM2, IFI16 and ZBP1/DLM1 (Barber, 2011).
With regards to the mechanisms that underlie PRR activation, a key remaining question is whether MAMP and DAMP detection is a direct ligand–receptor interaction or whether it requires multiple cofactors and complex modifications of host metabolic and homeostatic functions. In support of the latter hypothesis, numerous lines of evidence have recently highlighted the importance of mitochondria and mitochondrial functions in the regulation of host innate immune signalling. Importantly, although this organelle seems to be a signalling hub for innate immunity, there is strong compelling evidence to show that not only do mitochondria provide a molecular platform for signalling, but also that innate immunity and basic mitochondrial functions are integrated. Therefore, an emerging concept is that innate immune signalling is under the functional check of basic host metabolic functions, such as oxygen consumption, ATP production and possibly biosynthetic pathways that depend on mitochondrial activity and fitness.
PAMP detection and glycolytic switch
The main source of energy in the cell is the ATP that is generated through mitochondrial oxidative phosphorylation or through glycolysis. Oxidative phosphorylation is active in the presence of oxygen, whereas glycolysis is favoured under anaerobic conditions. The switch towards glycolytic ATP has recently been implicated in the activation and maturation of murine dendritic cells (Jantsch et al, 2008; Krawczyk et al, 2010). Dendritic cells stimulated with various TLR agonists have increased lactate production, higher expression of glucose transporter 1 and decreased oxygen consumption, all of which are signals of glycolytic metabolism (Krawczyk et al, 2010). Importantly, LPS‐induced glycolysis is dependent on the PI3K/AKT pathway and could be negatively regulated by the anti‐inflammatory cytokine IL‐10 or AMP‐activated protein kinase, a key regulator of oxidative phosphorylation (Krawczyk et al, 2010). In addition, macrophages are also highly dependent on glycolysis for ATP production, which is elicited by the transcription factor HIF1α—a key regulator of hypoxia (Cramer et al, 2003). Although a rapid increase in ATP through glycolysis is important for macrophage function, there is evidence to suggest that glycolysis can also be a protective mechanism against inflammatory stimuli (Garedew et al, 2010). LPS and IFNγ stimulation normally result in nitric‐oxide‐dependent mitochondrial dysfunction, leading to a collapse of the mitochondrial membrane potential (Δψm) and apoptosis (Garedew et al, 2010). Interestingly, LPS and IFNγ‐activated macrophages are less vulnerable as a result of glycolytic ATP production, which can maintain Δψm, thereby preventing cellular apoptosis (Garedew et al, 2010). The regulation of cellular metabolism by switching from oxidative phosphorylation to glycolysis seems to be critical in the activation and protection of certain immune cell populations.
Mitochondrial targeting by microbial effectors
Mitochondria are known to control intrinsic apoptotic cell death, and as host‐cell death is a common feature of infected cells, early reports focused on a possible impact of microbial‐derived proteins on host‐cell viability (Arnoult et al, 2009a). Indeed, several viruses have been shown to block mitochondria‐dependent apoptotic cell death, including those that encode viral homologues of the anti‐apoptotic protein BCL2, such as the 19 kDa product of the adenovirus E1B gene (named E1B‐19K) (Degenhardt et al, 2000) or the Epstein–Barr virus protein BHRF1 (Henderson et al, 1993). Other viral proteins, such as F1L and N1L proteins from vaccinia virus and vMIA from human cytomegalovirus, block apoptosis by interfering with the pro‐apoptotic proteins BAK and BAX at the mitochondrial outer membrane (Kvansakul et al, 2007; Wasilenko et al, 2003). Another group of viruses instead seems to promote host‐cell apoptosis as a pathogenic mechanism. This is the case of the viral protein R of human immunodeficiency virus 1, which interacts with the mitochondrial outer membrane protein VDAC, resulting in the opening of the mitochondrial permeability transition pore (mPTP), loss of the inner membrane potential Δψm and cytosolic leakage of host apoptogenic factors (Jacotot et al, 2000). The hepatitis B virus protein HBx induces similar mitochondrial collapse after interacting with VDAC3, leading to massive mitochondrial fragmentation and aggregation, and rapid apoptotic cell death (Rahmani et al, 2000; Table 1).
Table 1 Viral and bacterial effectors that target mitochondria
Several bacterial effectors also target mitochondria to manipulate host‐cell death and/or survival pathways. Enteropathogenic Escherichia coli proteins EspF and Map have amino‐terminal mitochondrial addressing sequences, and targeting of this organelle induces Δψm loss and cell death (Nagai et al, 2005); the specific host mitochondrial proteins that interact with EspF and Map remain unknown. In addition, several bacterial toxins target mitochondria and induce cell death. This is the case of VacA from Helicobacter pylori, Staphylococcus aureus α‐toxin and Clostridium difficile toxin A and B (Kozjak‐Pavlovic et al, 2008). Shigella flexneri also induces cell death in non‐myeloid cells through a mechanism that depends on the opening of the mPTP pore, resulting in Δψm loss and necrotic cell death, but whether this process depends on the injection of a specific bacterial type III secretion system effector remains unknown (Carneiro et al, 2009). Finally, Legionella pneumophila effector protein SdhA promotes survival of the infected cells by counteracting necrotic cell death as a result of mPTP opening (Laguna et al, 2006; Table 1).
Mitochondrial dynamics and antiviral immunity
On infection, viruses are rapidly detected by the innate immune system through several classes of PRR, including TLRs and RLRs, which recognize viral components and directly activate immune cells. On activation, TLRs recruit adaptor proteins such as MyD88 and TRIF, leading to downstream signalling cascades and production of proinflammatory cytokines such as type I interferons (IFNs) and chemokines (Akira & Takeda, 2004). Viral RNA that is synthesized in the cytoplasm of the cell or that is present in viral genomes already released into the cells is not accessible to TLR3, TLR7 or TLR8, as these TLRs detect viral nucleic acids presented on the extracellular or luminal side of host membranes, such as endosomal compartments (Akira & Takeda, 2004). Furthermore, fibroblasts and dendritic cells that lack MyD88 and TRIF can produce type I IFN after infection with a number of viruses, suggesting that the TLR system is not always crucial for innate immune defences against viral infection (Kato et al, 2005).
A new pathway of TLR‐independent responses to viruses was uncovered with the discovery of RIGI, the founding member of the RLR family (Takeuchi & Akira, 2010; Yoneyama et al, 2004), which also includes MDA5. These are cytosolic helicases that contain two CARD domains (Takeuchi & Akira, 2010; Yoneyama & Fujita, 2008) and sense viral RNA (Yoneyama & Fujita, 2008). Both helicases acquire ATPase activity by binding to their ligands, which is required to induce the conformational changes that lead to the exposure of the CARDs that are otherwise masked by the carboxy‐terminal regulatory domain. This conformational change is required for a putative interaction with the CARD domain of the mitochondrial adaptor MAVS (also known as IPS1, Cardif or VISA; Kawai et al, 2005; Meylan et al, 2005; Seth et al, 2005; Xu et al, 2005). MAVS, through the recruitment of TRAF3 and TRAF6, then activates two cytosolic protein kinase complexes: the ‘non‐canonical’ IKK‐related kinase TBK1 or IKK‐i/e associated with various adaptor proteins, such as TANK, NAP1 and NEMO; or one comprising IKKα, IKKβ and NEMO (Takeuchi & Akira, 2010). The TBK1 complex induces the phosphorylation and dimerization of the transcription factors IRF3 and IRF7, which translocate to the nucleus and bind to IFN‐stimulated response elements (ISREs), thereby resulting in the expression of type I IFN genes and a set of IFN‐inducible genes. The IKK complex activates NF‐κB, subsequently promoting the expression of pro‐inflammatory cytokines (Takeuchi & Akira, 2010; Fig 1).
Figure 1
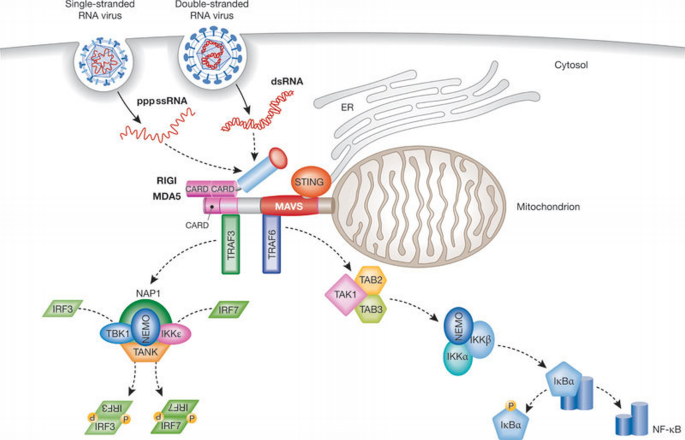
Overview of the RIGI‐like receptor pathway. Viral RNAs are recognized in the cytosol by the helicases RIGI or MDA5. The amino‐termini of RIGI and MDA5 contain two CARD domains that interact with the CARD domain of the mitochondrial adaptor MAVS. MAVS, after recruitment of transactivators, induces phosphorylation of IRF3 and IRF7 and NF‐κB activation, leading to the production of type I IFNs and pro‐inflammatory cytokines, respectively. dsRNA, double‐stranded RNA; ER, endoplasmic reticulum; IκBα, inhibitor of κ light polypeptide gene enhancer in B cells, α; IKKε, IκB kinase; IRF, interferon response factor; MDA5, melanoma differentiation associated gene 5; NAP, NF‐κB‐activating kinase‐associated protein; NEMO, NF‐κB essential modifier; NF‐κB, nuclear factor κB; pppssRNA, 5′ triphosphate single stranded RNA; RIGI, retinoic acid‐inducible gene I; STING, stimulator of interferon genes; TAB, TAK1‐associated binding protein; TAK, TGF‐β‐activated kinase; TANK, TRAF family member‐associated NF‐κB activator; TBK, TANK‐binding kinase; TRAF, TNF‐receptor‐associated factor.
The failure of MAVS‐deficient mice to mount a proper IFN response to Poly(I:C) stimulation and their severely compromised immune defence against infections with RNA viruses are evidence for the essential role of MAVS in antiviral innate immunity (Kumar et al, 2006; Sun et al, 2006). Nevertheless, how MAVS‐mediated signalling is engaged is not well understood. A recent study unveiled a crucial role of ubiquitin chains in IRF3 activation downstream from MAVS through the ubiquitin‐binding domains of NEMO (Zeng et al, 2009). In this model, NEMO is a sensor of Lys 63 polyubiquitin chains that activate TBK1 (Fig 2). However, the E3 ubiquitin ligase or ligases that mediate IRF3 activation by MAVS remain to be identified. A candidate is TRAF3, because it has been shown to be important for type I IFN production by RLRs (Oganesyan et al, 2006; Saha et al, 2006). However, in some models, the knockdown of TRAF3 does not impair the activation of the IFNβ promoter (Zeng et al, 2009), suggesting that other E3 ubiquitin ligases can compensate for the loss of TRAF3.
Figure 2
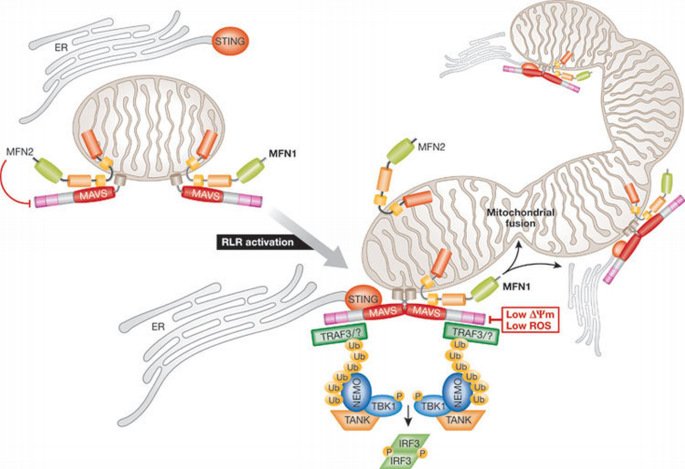
Mitochondrial dynamics and innate immunity. In the mitochondrial outer membrane, MAVS interacts with both MFN1 and MFN2. MFN2 is a direct MAVS inhibitor. RLR activation induces MAVS oligomerization, which recruits TRAF3 and other E3 ubiquitin ligases that catalyse Lys 63 polyubiquitination of target proteins, including TRAF3. Polyubiquitin chains recruit TANK and NEMO, which in turn bind and activate TBK1, leading to the phosphorylation of IRF3. Simultaneously, MFN1 leads to the redistribution of MAVS along mitochondria and a fusion of the mitochondrial network that promotes the interaction between MAVS and STING. Low Δψm or decreased ROS inhibit MAVS‐mediated signalling. Δψm, mitochondrial membrane potential; ER, endoplasmic reticulum; IRF, interferon response factor; MAVS, mitochondria‐associated viral sensor; MFN, mitofusin; NEMO, NF‐κB essential modifier; RLR, RIG‐I‐like receptor; ROS, reactive oxygen species; STING, stimulator of interferon genes; TANK, TRAF family member‐associated NF‐κB activator; TBK, TAK1‐associated binding protein; TRAF, TNF‐receptor‐associated factor.
Importantly, MAVS must be localized to mitochondria to exert its function (Seth et al, 2005), suggesting that the mitochondrial environment is required for signal transduction. In line with this, two independent studies showed that MFN1—an effector of the mitochondrial fusion machinery (Chan, 2006)—positively regulates MAVS‐mediated innate antiviral responses (Castanier et al, 2010; Onoguchi et al, 2010). In both cases, MAVS was shown to interact with MFN1: after RLR activation, the complex MFN1–MAVS regulates either MAVS redistribution along mitochondria (Onoguchi et al, 2010) or the elongation of the mitochondrial network (Castanier et al, 2010). Although the role of MAVS redistribution remains to be defined, it has been proposed that the elongation of the mitochondrial network linked to RLR activation enhances the association of MAVS with STING (Castanier et al, 2010)—an antiviral signalling adaptor localized in the endoplasmic reticulum membrane (Ishikawa & Barber, 2008)—therefore amplifying the antiviral response (Fig 1). Surprisingly, although MFN1 seems to be a positive regulator in the RLR pathway (Castanier et al, 2010; Onoguchi et al, 2010), its close homologue MFN2 is a direct inhibitor of MAVS, a function that is probably unrelated to its role in mitochondrial dynamics (Yasukawa et al, 2009). Thus, although MFN1 and MFN2 have a similar function in mitochondrial fusion (Chan, 2006), it seems that they have opposite roles in viral innate immunity. As MFN1 and MFN2 both engage in homotypic and heterotypic interactions on the mitochondrial outer membrane, associations with their respective partners might ensure fine‐tuning of MAVS‐mediated signalling, in addition to the regulation of mitochondrial fusion.
MAVS‐mediated antiviral signalling is not only regulated by mitochondrial dynamics. Two mitochondrial proteins, NLRX1 (Moore et al, 2008) and gC1qR (Xu et al, 2009), have been recently identified as regulators of MAVS function that are involved in the inhibition of the RIGI‐ and MDA5‐dependent antiviral response. The autophagy‐related protein ATG5/12 (Jounai et al, 2007) and several other cellular proteins such as PCBP2/AIP4 (You et al, 2009), RNF5 (Zhong et al, 2010) and PLK1 (Vitour et al, 2009) have also been implicated in the negative regulation of MAVS‐mediated signalling. Interestingly, a recent study has reported that Δψm is crucial for this pathway (Koshiba et al, 2011). Cells in which the Δψm was dissipated—treated with the protonophore CCCP or lacking both MFN1 and MFN2—are defective in the antiviral innate immune response after RLR, but not TLR3, activation. The precise role of Δψm in MAVS‐mediated signalling remains to be determined, although Koshiba and colleagues suggest that loss of Δψm might prevent the structural rearrangement of the MAVS complex, which could exist as a readily available pool on the mitochondrial membrane to provide a quicker response to viral infection (Koshiba et al, 2011). Furthermore, loss of Δψm is associated with an intensive fragmentation of the mitochondrial network that might prevent the association of MAVS with the endoplasmic‐reticulum‐associated antiviral signalling adaptor STING (Castanier et al, 2010; Ishikawa & Barber, 2008). Importantly, another conclusion of this study is that the inhibition of ATP synthesis—one of the most prominent functional role of mitochondria—does not impair MAVS‐mediated signalling.
An indirect role of mitochondria in the regulation of innate immune responses is shown by the fact that ROS modulate several signalling pathways, including the NF‐κB and JNK pathways, and the caspase 1 inflammasome (Naik & Dixit, 2011). The two main sources of cellular ROS are the mitochondria and the membrane‐associated NADPH oxidase (NOX) and dual oxidase (DUOX) complexes. NOX2 and ROS were recently shown to be required for the triggering of efficient MAVS‐mediated antiviral signalling (Soucy‐Faulkner et al, 2010). The authors also provide evidence that NOX2 is crucial for the expression of MAVS because NOX2 downregulation diminishes MAVS mRNA expression without affecting its mitochondrial localization (Soucy‐Faulkner et al, 2010).
Although the function of mitochondria in MAVS‐mediated antiviral signalling warrants further investigation because mitochondria are essential for MAVS function (Seth et al, 2005), recent studies have suggested that mitochondrial dynamics, Δψm and ROS are crucial regulators and/or effectors of this pathway (Fig 2).
Mitochondria: central platform for innate immunity
The identification of MAVS as a central adaptor protein of the RLR pathway that localizes to the mitochondrial outer membrane provided the first example of a functional link between mitochondrial function and innate immune signalling. Notably, recent evidence demonstrates that many host defence pathways are controlled by this organelle (Fig 3).
Figure 3
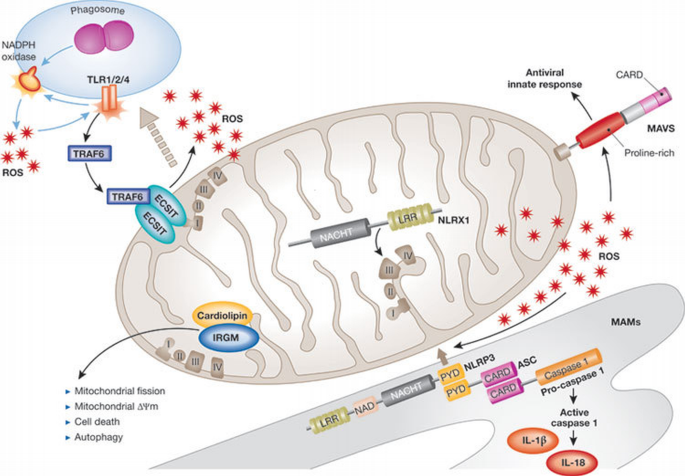
Innate immune signalling at the mitochondria. On induction of mitochondrial reactive oxygen species (ROS), NLRP3 localizes to the mitochondrial‐associated endoplasmic reticulum membrane (MAM) with ASC and pro‐caspase 1, inducing caspase‐1 activation and the production of IL‐1β and IL‐18. MAVS is located on the outer mitochondrial membrane and mediates antiviral signalling. On TLR1/2/4 stimulation, TRAF6 translocates to the mitochondria and interacts with ECSIT, contributing to ROS production. NLRX1 is internalized into the mitochondrial matrix where it interacts with UQCRC2, a protein of the complex III of the electron transfer chain, potentiating ROS production. IRGM binds to the mitochondrial lipid cardiolipin, influencing autophagy, mitochondrial fission, mitochondrial Δψm and cell death. ASC, apoptosis‐associated speck‐like protein containing a CARD; ECSIT, evolutionarily conserved signalling intermediate in Toll pathway; IL, interleukin; IRGM, immunity‐related GTPase family M protein; LRR, leucine‐rich repeat; MAVS, mitochondria‐associated viral sensor; NAD, nicotinamide adenine dinucleotide; NLRP, NOD‐like receptor family, pyrin domain containing; NLRX, NOD‐like receptor family member X; PYD, pyrin domain; TLR, Toll‐like receptor; TRAF, TNF‐receptor‐associated factor.
NLRP3. NLRP3 is a crucial NLR protein implicated in the induction of the caspase 1 inflammasome in response to a surprisingly wide spectrum of microbial‐associated and danger‐associated molecular signatures (see Benko et al (2008) for a recent comprehensive review on this topic). The pleiotropic nature of the upstream activators of NLRP3 has led to speculation that NLRP3 senses a common cellular signal triggered by PAMPs and DAMPs rather than several individual molecular entities. The nature of this common signal is still open to debate, as evidence suggests that either lysosomal damage or ROS could act as NLRP3 activators (Tschopp & Schroder, 2010). Although phagosome‐associated ROS generated by the NADPH complex were first proposed to trigger NLRP3 responses, these observations have been challenged by the fact that normal NLRP3 responses occur in cells carrying mutations that impair NADPH‐oxidase‐dependent ROS production (van Bruggen et al, 2010). The group of Jürg Tschopp recently proposed that mitochondria could be the main source of ROS contributing to NLRP3 activation (Zhou et al, 2011), providing a possible explanation for these conflicting results. They found that rotenone and antimycin—two drugs that block the mitochondrial respiratory chain and lead to the generation of mitochondrial ROS—potently activate NLRP3. Similarly, inhibition of autophagy results in the accumulation of defective and ROS‐producing mitochondria, which potentiates NLRP3‐dependent inflammasome activation. Interestingly, the authors also demonstrated that mitochondria act as a molecular scaffolding platform for the recruitment of several members of the NLRP3 inflammasome—including NLRP3 itself and the adaptor protein ASC—in response to known NLRP3 activators, such as monosodium urate, aluminium hydroxide and nigericin (Zhou et al, 2011). The mechanism that underlies NLRP3 inflammasome recruitment to the vicinity of ROS‐producing mitochondria remains unknown, and future studies are necessary to identify the mitochondria‐associated docking complexes for NLRP3 inflammasome components.
TLRs. The engulfment of bacteria by phagocytic cells results in the activation of numerous innate immune signalling pathways, including those that promote microbicidal activity. ROS generation is one of the most ancient and efficient means to kill microorganisms and it is functionally linked to phagocytosis through the upregulation of the NADPH oxidase complex, which is localized at the surface of the phagosome. TLR signalling enhances NADPH oxidase activity and, in turn, ROS generated by this complex potentiates TLR‐mediated induction of inflammatory pathways, such as those dependent on NF‐κB and MAP kinases (Gill et al, 2010). A recent study showed that mitochondria might also have crucial roles in phagosome‐associated ROS generation and in the connection to TLR signalling. Indeed, activation of murine macrophages with ligands of cell‐surface TLRs—TLR1, TLR2 and TLR4—but not of endosomal TLRs—TLR3, TLR7, TLR8 and TLR9—resulted in the production of mitochondria‐derived ROS (West et al, 2011). Interestingly, when cells phagocytosed latex beads coated with TLR2 ligand Pam3CSK4 or TLR4 ligand LPS, mitochondria accumulated near to phagosomes containing the latex beads, an effect that was not observed if beads were coated with the endosome‐associated TLR9 ligand CpG DNA. The authors concluded that engulfment of microbes, concomitant with TLR‐mediated detection, resulted in mitochondrial accumulation around phagosomes and local production of ROS, which could contribute to the elimination of the engulfed microorganism. TRAF6—an E3 ligase crucial for TLR signalling—is translocated to the mitochondria in response to the engagement of TLR1/2/4 ligands, but not TLR3/9 ligands, and mitochondrial ECSIT protein was identified as a TRAF6‐dependent target of ubiquitination after macrophage activation by TLR1/2/4 ligands. Importantly, triggering of the TRAF6–ECSIT pathway at the mitochondria was responsible for the recruitment of these organelles to the vicinity of phagosomes and the induction of mitochondrial ROS. Therefore, this study highlights the unexpected importance of mitochondrial ROS generation in phagosomal killing, and provides a new example of how mitochondria can act as a signalling hub—in this case, the TRAF6–ECSIT pathway—for innate immune signalling (West et al, 2011).
NLRX1. NLRX1 is a member of the NLR family that has a unique N‐terminal domain—which accounts for the letter ‘X’ in its acronym—that was found to contain a mitochondrial addressing sequence (Moore et al, 2008; Tattoli et al, 2008). Biochemical analyses demonstrated that NLRX1 localizes to the mitochondrial matrix (Arnoult et al, 2009b). Strikingly, NLRX1 represents the first and so far only example of a PRR family member that targets this cellular location, therefore implying that NLRX1 probably establishes a fundamental link between mitochondrial functions and innate immunity. However, the fact that NLRX1 targets the mitochondrial matrix is puzzling, as this confined location makes it unlikely that the protein would directly detect microbial‐derived molecular signatures in an archetypal manner. Thus, the exact function of NLRX1 is unclear. Overexpression of NLRX1 was shown to induce ROS production (Tattoli et al, 2008), which can be explained by its interaction with UQCRC2, a matrix‐facing component of the mitochondrial respiratory chain complex III that has a crucial role in ROS generation (Arnoult et al, 2009b). In _Chlamydia_‐infected cells, NLRX1 potentiates the ROS production triggered by the pathogen, an effect that is required for optimal bacterial growth (Abdul‐Sater et al, 2010). Interestingly, NLRX1 was also found to interact with MAVS, the crucial adaptor protein of RLR pathways, on the cytosolic side of the mitochondrial outer membrane, resulting in inhibition of MAVS‐dependent antiviral responses (Moore et al, 2008). However, how NLRX1 interacts with MAVS remains unclear because these two proteins are targeted to distinct mitochondrial subdomains that are separated by two membranes. As NLRX1 entry into mitochondria was shown to depend on the inner membrane potential Δψm (Arnoult et al, 2009b), it is conceivable that dissipation of Δψm would result in the retention of NLRX1 in the cytosol where the protein could interact with and inhibit MAVS‐dependent signalling. This hypothesis has not been tested experimentally and it would be important to determine whether viral infection could induce cytosolic retention of NLRX1. Alternatively, NLRX1 could influence RLR signalling through ROS production, which has been shown to modulate MAVS‐dependent pathways (Soucy‐Faulkner et al, 2010).
IRGM. IRGM is a ubiquitously expressed human guanosine triphosphatase (Hunn et al, 2011) that functions in the innate immune control of intracellular pathogens. In contrast to the many murine IRG genes that are regulated by interferons, only two orthologues exist in humans—IRGM and IRGC. IRGM was initially thought to be a pseudogene but has a role in the elimination of M. tuberculosis in human macrophages (Singh et al, 2006). Specifically, IRGM was demonstrated to be crucial in rapamycin‐, starvation‐ and IFNγ‐induced autophagy to restrict the intracellular growth of M. tuberculosis in infected human macrophages (Singh et al, 2006). Importantly, single nucleotide polymorphisms in IRGM have been identified as risk factors for Crohn's disease (Parkes et al, 2007) and susceptibility to tuberculosis (Intemann et al, 2009) in different human populations. The mechanism of IRGM‐mediated autophagy was largely unknown, but a recent study from the Deretic group has revealed the importance of mitochondria in this process. IRGM localizes to mitochondria and regulates mitochondrial fission events, which had been previously implicated in autophagic control of intracellular pathogens (Singh et al, 2010). The localization of IRGM to mitochondria is dependent on the interaction of IRGM with cardiolipin, a mitochondrial lipid found predominately in the inner mitochondria membrane. Additionally, a detailed analysis of IRGM spliced variants indicated that preferential overexpression of IRGMd isoform induces mitochondrial fission, mitochondrial depolarization and autophagy‐independent cell death, leading to the release of HMGB1, a major pro‐inflammatory alarmin/DAMP (Singh et al, 2010). Altogether, the dynamic regulation of mitochondria seems to be a central aspect of the dual function of IRGM in mediating autophagy (cell survival) and apoptosis and/or necrosis (cell death) in innate immunity. Future studies need to explore the role of intra‐mitochondrial IRGM in the context of bacterial infection.
Modulation of innate immunity by mDAMPs
Mitochondria probably evolved from a bacterial ancestor that established a symbiotic relationship with its host cell and, in this regard, mitochondrial DAMPs (herein termed ‘mDAMPs’) can be viewed as intermediate entities between DAMPs and PAMPs. Not surprisingly, mitochondria have several unique features in the cell and several mitochondrial elements—such as cytochrome c, formyl peptides and mitochondrial DNA (mtDNA)—seem to have been selected during evolution to act as mDAMPs that alert the cell to damage in the mitochondria. By far the most studied mDAMP is cytochrome c, which triggers apoptotic cell death after its release from the mitochondrial inner membrane into the cytosol, where it interacts with APAF1 to trigger the activation of the caspase‐9‐dependent pathways of apoptosis. Although mitochondria‐dependent apoptotic cell death is a crucial aspect of innate immune responses to pathogens, it has been extensively reviewed elsewhere (Garrido et al, 2006) and is not discussed here. Instead, we focus on more recent aspects of the link between mDAMP liberation and innate immunity (Fig 4).
Figure 4
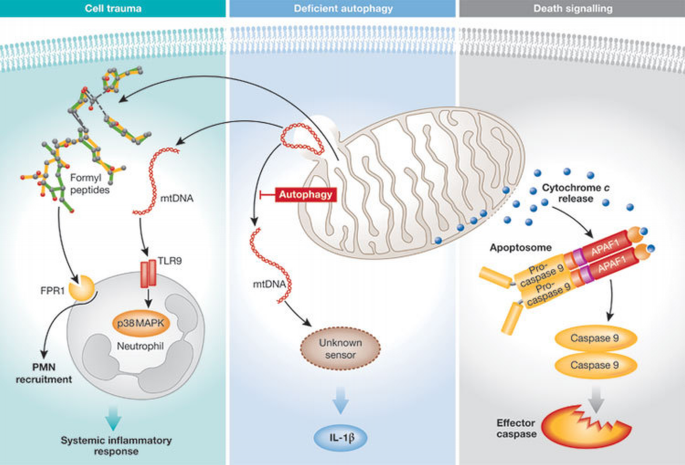
Release of mitochondrial DAMPs triggers cell death and inflammatory pathways. (A) Severe trauma can induce the release of mitochondrial DNA (mtDNA), which is detected by TLR9 located on the neutrophil surface, and thus activates p38 MAP kinase. N‐formyl peptides are released by mitochondria and recruit polymorphonuclear neutrophils (PMNs) by binding to the formyl‐peptide receptor 1 (FPR1) expressed on their surface. (B) Autophagy‐deficient cells accumulate defective mitochondria, which release mtDNA into the cytosol, triggering activation of the inflammasome through an undefined cytosolic sensor. (C) Cell death signals induce the release of cytochrome c into the cytosol, leading to the recruitment of APAF1 and pro‐caspase 9 into a multiprotein complex known as the apoptosome. After activation, caspase 9 induces apoptosis by cleaving effector caspases. DAMP, danger‐associated molecular pattern; IL, interleukin; MAPK, mitogen‐activated protein kinase; TLR, Toll‐like receptor.
Circulating mDAMPs in trauma. Zhang and colleagues hypothesized that, in trauma patients and organ injuries without an open wound, cellular damage would lead to the release of mitochondrial molecules that could operate as mDAMPs and initiate an inflammatory response (Zhang et al, 2010a). The authors noticed a considerable increase in the concentration of mtDNA in the circulation of trauma patients as compared with healthy volunteers. MtDNA released at the site of the injury recruited polymorphonuclear neutrophils (PMNs), which were also activated through a TLR9‐dependent pathway. In addition to mtDNA, mitochondrial formyl peptides were also proposed to act as mDAMPs during injury. Stimulation of the G‐protein‐coupled receptors FPR1 and FPRL1—which mediate the chemotactic and stimulatory effects of N‐formyl‐Met‐Leu‐Phe (fMLP)—was shown to occur on PMNs after exposure to mDAMPs. This indirectly suggests that endogenous mitochondrial formyl peptides are liberated in vivo at the site of injury and in the circulation. Therefore, liberation of mDAMPs can account for the induction of sterile inflammation after organ injury and in trauma patients, which can result in a systemic inflammatory response syndrome similar to that observed during septic shock.
MtDNA and inflammasome activation. There has recently been increasing interest in the identification of intracellular sensors of foreign DNA (Barber, 2011), and type I IFN induction and inflammasome activation seem to represent two of the main host responses to intracellular DNA detection. Several PRRs have been shown to mediate the host response to intracellular DNA: ZBP1/DLM1 is crucial for the induction of type I IFN in some cell types, whereas in others the conversion of DNA into RNA by the RNA polymerase III has been shown to redirect host responses to the RIGI/MAVS pathway (Barber, 2011). However, the physiological relevance of the latter system in the context of bacterial infection has been challenged (Monroe et al, 2009). Recently, the PYHIN protein IFI16 (p204 in mice) was also shown to detect cytosolic DNA, leading to the induction of type I IFN after recruitment and activation of STING, which seems to act as a key adaptor molecule for several intracellular DNA‐sensing systems (Unterholzner et al, 2011). In addition, activation of the caspase 1 inflammasome in response to foreign DNA requires AIM2, another cytosolic PYHIN protein (Barber, 2011).
MtDNA can be released from damaged mitochondria (Zhang et al, 2010b) and—as mtDNA has common features with bacterial genomes—the potential role of mtDNA in the activation of host responses to cytosolic DNA is starting to be analysed. The inhibition of autophagy results in the accumulation of defective mitochondria and the cytosolic release of mtDNA (Nakahira et al, 2011). Interestingly, the presence of cytosolic mtDNA—and not the accumulation of damaged mitochondria—was shown to induce activation of the caspase 1 inflammasome in a partly AIM2‐independent manner. This suggests that an unknown cytosolic sensor of mtDNA might contribute to the activation of the inflammasome.
Concluding remarks
Accumulating evidence has introduced the concept that mitochondria—in addition to their well‐known role in cell metabolic processes and cell death—are crucial assembly platforms for innate immune signalling, as summarized in this review. Among the outstanding questions that remain poorly understood (Sidebar A) is: why mitochondria? An attractive possibility is that the convergence of multiple innate immune pathways on this organelle would allow for a tight functional integration of host defence and metabolic processes, although this model has not yet been thoroughly tested. Could it be that the activation of certain innate immune responses requires transition through an ‘energetic checkpoint’? This could ensure that efficient cell‐intrinsic defence pathways would only be turned on in cells that remain fit. Another probable reason for the functional coupling of some innate immune pathways to mitochondria is that this organelle is a major cellular source of ROS, which contribute in many ways to host defence against pathogens. The topological organization of host defence pathways at the mitochondria might therefore have evolved because ROS are intrinsically unstable molecules that cannot diffuse into the cell.
Sidebar A | In need of answers
- i.
Why does MAVS have to be anchored to the mitochondrial outer membrane to exert its function? - ii.
What is the precise role of Δψm in MAVS‐mediated signalling? - iii.
Do some BCL2 family members also regulate MAVS‐mediated innate immunity? - iv.
Which signals activate NLRX1 in the mitochondrial matrix? - v.
To what extent do mitochondrial metabolic functions have an impact on the innate immune pathways that require this organelle as a signalling hub? - vi.
What is the role of mitochondria in innate immunity in other organisms, such as Drosophila?
Mitochondria have been studied for decades regarding their role in cellular metabolism. Equally as unexpected as the discovery 20 years ago that this organelle controls programmed cell death, the mitochondrion now emerges as a key component in host innate immune defence pathways. The implications of this new paradigm are only starting to emerge.
Δψm:
mitochondrial membrane potential
AIM2:
absent in melanoma 2
AKT:
cellular homologue of v‐AKT
ASC:
apoptosis‐associated speck‐like protein containing a CARD
ATG:
autophagy‐specific gene
BAK:
BCL2‐antagonist/killer
BAX:
BCL2‐associated X protein
BCL2:
B‐cell leukaemia/lymphoma 2
ECSIT:
evolutionarily conserved signalling intermediate in Toll pathway
FPR1:
formyl peptide receptor 1
FPRL1:
formyl peptide receptor‐like 1
gC1q:
complement component 1, q subcomponent binding protein
HIF1α:
hypoxia inducible factor 1, α‐subunit
IFI16:
interferon, γ‐inducible protein 16
IFNγ:
interferon γ
IKK:
IκB kinase
IL‐10:
interleukin 10
IRF:
interferon response factor
IRGM:
immunity‐related GTPase family M protein
LPS:
lipopolysaccharide
MAVS:
mitochondria‐associated viral sensor
MDA5:
melanoma differentiation associated gene 5
MFN1:
mitofusin 1
MyD88:
myeloid differentiation primary response gene 88
NAP1:
NF‐κB‐activating kinase‐associated protein 1
NEMO:
NF‐κB essential modifier
NLRX1:
NOD‐like receptor family member X1
NOX2:
NADPH oxidase 2
PCBP2/AIP4:
poly(rC) binding protein 2 / atrophin 1 interacting protein 4
PI3K:
phosphoinositide‐3‐kinase
PLK1:
polo‐like kinase 1
Poly (I:C):
polyinosine‐polycytidylic acid
RIGI:
retinoic acid‐inducible gene I
RNF5:
RING finger protein 5
ROS:
reactive oxygen species
STING:
stimulator of interferon genes
TANK:
TRAF family member‐associated NF‐κB activator
TBK1:
TANK‐binding kinase 1
TRAF:
TNF‐receptor‐associated factor
TRIF:
TIR‐domain containing adaptor inducing IFN‐β
UQCRC2:
ubiquinol‐cytochrome c reductase core protein II
VDAC:
voltage‐dependent anion channel
ZBP1/DLM1:
Z‐DNA binding protein 1
References
- Abdul‐Sater AA et al (2010) Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod‐like family member NLRX1. J Biol Chem 285: 41637–41645
Google Scholar - Akira S, Takeda K (2004) Toll‐like receptor signalling. Nat Rev Immunol 4: 499–511
Google Scholar - Arnoult D, Carneiro L, Tattoli I, Girardin SE (2009a) The role of mitochondria in cellular defense against microbial infection. Semin Immunol 21: 223–232
Google Scholar - Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE (2009b) An N‐terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci 122: 3161–3168
Google Scholar - Barber GN (2011) Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol 23: 10–20
Google Scholar - Benko S, Philpott DJ, Girardin SE (2008) The microbial and danger signals that activate Nod‐like receptors. Cytokine 43: 368–373
Google Scholar - Carneiro LA et al (2009) Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5: 123–136
Google Scholar - Castanier C, Garcin D, Vazquez A, Arnoult D (2010) Mitochondrial dynamics regulate the RIG‐I‐like receptor antiviral pathway. EMBO Rep 11: 133–138
Google Scholar - Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252
Google Scholar - Cramer T et al (2003) HIF‐1alpha is essential for myeloid cell‐mediated inflammation. Cell 112: 645–657
Google Scholar - Degenhardt K, Perez D, White E (2000) Pathways used by adenovirus E1B 19K to inhibit apoptosis. Symp Soc Exp Biol 52: 241–251
Google Scholar - Elinav E, Strowig T, Henao‐Mejia J, Flavell RA (2011) Regulation of the antimicrobial response by NLR proteins. Immunity 34: 665–679
Google Scholar - Fritz JH, Ferrero RL, Philpott DJ, Girardin SE (2006) Nod‐like proteins in immunity, inflammation and disease. Nat Immunol 7: 1250–1257
Google Scholar - Garedew A, Henderson SO, Moncada S (2010) Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ 17: 1540–1550
Google Scholar - Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13: 1423–1433
Google Scholar - Gill R, Tsung A, Billiar T (2010) Linking oxidative stress to inflammation: Toll‐like receptors. Free Radic Biol Med 48: 1121–1132
Google Scholar - Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A (1993) Epstein–Barr virus‐coded BHRF1 protein, a viral homologue of Bcl‐2, protects human B cells from programmed cell death. Proc Natl Acad Sci USA 90: 8479–8483
Google Scholar - Hunn JP, Feng CG, Sher A, Howard JC (2011) The immunity‐related GTPases in mammals: a fast‐evolving cell‐autonomous resistance system against intracellular pathogens. Mamm Genome 22: 43–54
Google Scholar - Intemann CD et al (2009) Autophagy gene variant IRGM ‐261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5: e1000577
Google Scholar - Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678
Google Scholar - Jacotot E et al (2000) The HIV‐1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med 191: 33–46
Google Scholar - Jantsch J et al (2008) Hypoxia and hypoxia‐inducible factor‐1 alpha modulate lipopolysaccharide‐induced dendritic cell activation and function. J Immunol 180: 4697–4705
Google Scholar - Jounai N et al (2007) The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 104: 14050–14055
Google Scholar - Kato H et al (2005) Cell type‐specific involvement of RIG‐I in antiviral response. Immunity 23: 19–28
Google Scholar - Kawai T, Akira S (2011) Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650
Google Scholar - Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988
Google Scholar - Koshiba T, Yasukawa K, Yanagi Y, Kawabata S (2011) Mitochondrial membrane potential is required for MAVS‐mediated antiviral signaling. Sci Signal 4: ra7
Google Scholar - Kozjak‐Pavlovic V, Ross K, Rudel T (2008) Import of bacterial pathogenicity factors into mitochondria. Curr Opin Microbiol 11: 9–14
Google Scholar - Krawczyk CM et al (2010) Toll‐like receptor‐induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115: 4742–4749
Google Scholar - Kumar H et al (2006) Essential role of IPS‐1 in innate immune responses against RNA viruses. J Exp Med 203: 1795–1803
Google Scholar - Kvansakul M, van Delft MF, Lee EF, Gulbis JM, Fairlie WD, Huang DC, Colman PM (2007) A structural viral mimic of prosurvival Bcl‐2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol Cell 25: 933–942
Google Scholar - Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR (2006) A _Legionella pneumophila_‐translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci USA 103: 18745–18750
Google Scholar - Loo YM, Gale M Jr (2011) Immune signaling by RIG‐I‐like receptors. Immunity 34: 680–692
Google Scholar - Medzhitov R (2007) Recognition of microorganisms and activation of the immune response. Nature 449: 819–826
Google Scholar - Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172
Google Scholar - Monroe KM, McWhirter SM, Vance RE (2009) Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog 5: e1000665
Google Scholar - Moore CB et al (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451: 573–577
Google Scholar - Nagai T, Abe A, Sasakawa C (2005) Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J Biol Chem 280: 2998–3011
Google Scholar - Naik E, Dixit VM (2011) Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 208: 417–420
Google Scholar - Nakahira K et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230
Google Scholar - Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G (2006) Critical role of TRAF3 in the Toll‐like receptor‐dependent and ‐independent antiviral response. Nature 439: 208–211
Google Scholar - Onoguchi K, Onomoto K, Takamatsu S, Jogi M, Takemura A, Morimoto S, Julkunen I, Namiki H, Yoneyama M, Fujita T (2010) Virus‐infection or 5'ppp‐RNA activates antiviral signal through redistribution of IPS‐1 mediated by MFN1. PLoS Pathog 6: e1001012
Google Scholar - Parkes M et al (2007) Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 39: 830–832
Google Scholar - Rahmani Z, Huh KW, Lasher R, Siddiqui A (2000) Hepatitis B virus X protein colocalizes to mitochondria with a human voltage‐dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol 74: 2840–2846
Google Scholar - Saha SK et al (2006) Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25: 3257–3263
Google Scholar - Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682
Google Scholar - Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313: 1438–1441
Google Scholar - Singh SB et al (2010) Human IRGM regulates autophagy and cell‐autonomous immunity functions through mitochondria. Nat Cell Biol 12: 1154–1165
Google Scholar - Soucy‐Faulkner A, Mukawera E, Fink K, Martel A, Jouan L, Nzengue Y, Lamarre D, Vande Velde C, Grandvaux N (2010) Requirement of NOX2 and reactive oxygen species for efficient RIG‐I‐mediated antiviral response through regulation of MAVS expression. PLoS Pathog 6: e1000930
Google Scholar - Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ (2006) The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24: 633–642
Google Scholar - Takeuchi O, Akira S (2009) Innate immunity to virus infection. Immunol Rev 227: 75–86
Google Scholar - Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820
Google Scholar - Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE (2008) NLRX1 is a mitochondrial NOD‐like receptor that amplifies NF‐kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep 9: 293–300
Google Scholar - Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10: 210–215
Google Scholar - Unterholzner L et al (2011) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11: 997–1004
Google Scholar - van Bruggen R, Koker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, van den Berg TK (2010) Human NLRP3 inflammasome activation is Nox1–4 independent. Blood 115: 5398–5400
Google Scholar - Vitour D et al (2009) Polo‐like kinase 1 (PLK1) regulates interferon (IFN) induction by MAVS. J Biol Chem 284: 21797–21809
Google Scholar - Wasilenko ST, Stewart TL, Meyers AF, Barry M (2003) Vaccinia virus encodes a previously uncharacterized mitochondrial‐associated inhibitor of apoptosis. Proc Natl Acad Sci USA 100: 14345–14350
Google Scholar - West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480
Google Scholar - Xu L, Xiao N, Liu F, Ren H, Gu J (2009) Inhibition of RIG‐I and MDA5‐dependent antiviral response by gC1qR at mitochondria. Proc Natl Acad Sci USA 106: 1530–1535
Google Scholar - Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell 19: 727–740
Google Scholar - Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, Kawabata S, Koshiba T (2009) Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2: ra47
Google Scholar - Yoneyama M, Fujita T (2008) Structural mechanism of RNA recognition by the RIG‐I‐like receptors. Immunity 29: 178–181
Google Scholar - Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5: 730–737
Google Scholar - You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z (2009) PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol 10: 1300–1308
Google Scholar - Zeng W, Xu M, Liu S, Sun L, Chen ZJ (2009) Key role of Ubc5 and lysine‐63 polyubiquitination in viral activation of IRF3. Mol Cell 36: 315–325
Google Scholar - Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010a) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107
Google Scholar - Zhang Q, Itagaki K, Hauser CJ (2010b) Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34: 55–59
Google Scholar - Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB (2010) The E3 ubiquitin ligase RNF5 targets virus‐induced signaling adaptor for ubiquitination and degradation. J Immunol 184: 6249–6255
Google Scholar - Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225
Google Scholar