Recessive PIEZO2 stop mutation causes distal arthrogryposis with distal muscle weakness, scoliosis and proprioception defects (original) (raw)
'Arthrogryposis' or arthrogryposis multiplex congenita (AMC) is a general descriptive term applied to a broad spectrum of diseases to describe congenital contractures of multiple joints.1 It is a symptom rather than a specific diagnosis, and further deciphering of the genetic basis is required.2, 3, 4
A currently 186/12-year-old, high school grade 3 student, with normal cognitive and academic performance presented at the age of 36/12 years with delay of independent ambulation and his development has been monitored since then (Figure 1, Supplementary Videos 1 and 2). He is the first and only child of 2nd degree consanguineous healthy Turkish parents. The parents, currently age 37 and 48 years, are healthy, have distal joint laxity without any complaints. The pregnancy was uneventful with normal intrauterine movements. The patient was born at term by vaginal delivery (birth weight: 3.5 kg; APGAR score: 4/6). He had hip dysplasia at birth and was hospitalized because of hypotonia but did not require ventilatory support. The infant had sucking and swallowing difficulties after birth, which required nasogastric feeding during the first 2 months of life. Our patient did not show facial weakness. Feeding difficulties persisted up to 4 years of age and nasal speech seemed to be due to weakness of oropharyngeal muscles. Evolution of symptoms and presentation with age may reflect a developmental maturational process. Head control was achieved at the age of 5 months and sitting without support at 8 months. The course of the disease was non-progressive with a mild improvement after the age of 6–7 years. Remarkably, he deteriorated after an upper respiratory infection, and required mechanical ventilation for a day at the age of 9 years in the intensive care unit. He started independent walking at the age of 16 years; at the time also he required surgery because of progressive scoliosis (right thoracal, left lumbar scoliosis 70°).
Figure 1
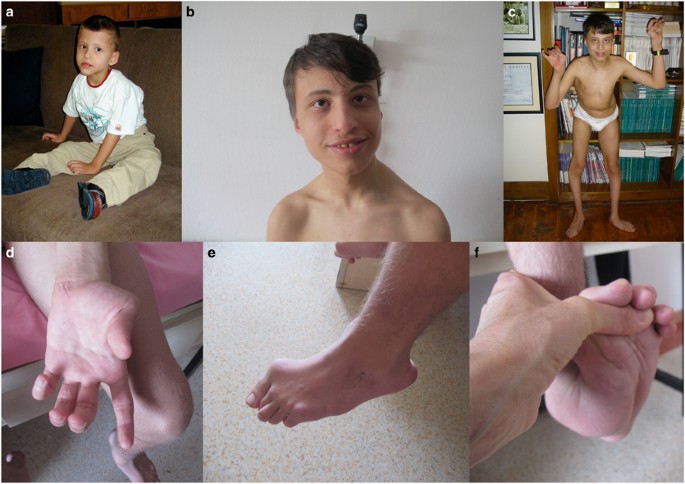
Clinical phenotype. Patient at the age of 5 years (a), 12 years (b) and 18 years (c–f). Note the elongated face, distal involvement, position of the hands and eversion deformity in the feet, scoliosis, wide-based unsteady gait; and distal laxity in upper and lower extremities.
His last follow-up at 186/12 years of age revealed an elongated face, atrophic phenotype, dysmorphic features, high-arched palate, marked distal involvement, distal laxity, abduction in hands (prominently thumb) and feet, flexion contractures, left sided scoliosis, asymmetric involvement of shoulder and trunk muscles, no facial involvement, rigid spine deformity, protruded calcaneus and eversion deformity of the feet. His body weight was 50 kg (<3p), and height 162 cm (<3p). He is able to walk eyes open, with a wide-based, unsteady gait and can stand up from the floor on his knees with support. Besides weakness, coordination problems were noted in the upper extremity and trunk muscles. Sensory examination demonstrated a proprioception defect prominent in the upper extremity; a Romberg test was not applicable by definition because of a wide-based gait, however there was worsening of balance and coordination when he closed his eyes. Joint position sense was lost in the lower extremity and was present at the level of the ankles. There was a mild left sided and upper extremity predominance. Pain, temperature, two-point discrimination and vibration senses were normal. The patient was inconsistent in sensing during the light touch examination.
At last follow-up he showed a slightly decreased lung function; a forced vital capacity of 73% was measured. Serum creatine kinase levels were normal. Echocardiography and electromyography at the age of 8 months showed physiological function. A muscle biopsy at the age of 36/12 years revealed mild myopathic changes with rounding of some fibers, mild variation in fiber size, few fibers with internal nuclei, focal increase in endomysial and perimysial connective tissue, and focal fatty tissue infiltration (Figure 2). Type I fibers were predominant and rare regenerating fibers were detected by neonatal myosin. A recent whole body muscle MRI showed nonspecific changes in the lower limbs (Supplementary Figure 1).
Figure 2
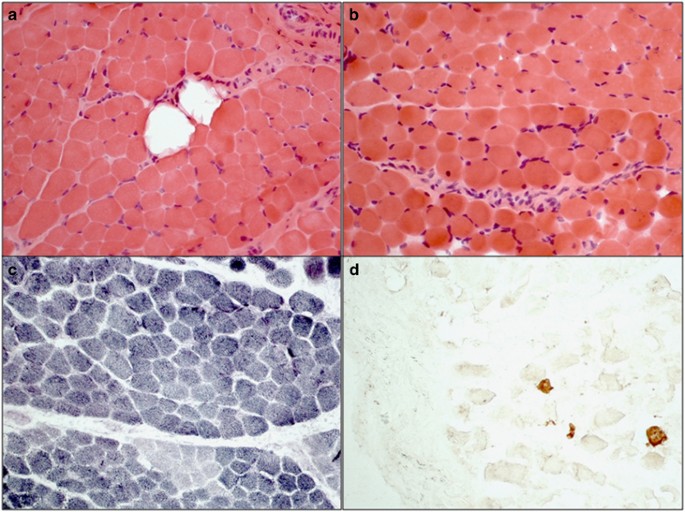
Muscle pathology. Muscle biopsy from the left M.vastus lateralis at 3.5 years of age showing variation in fiber size, mild increase in endomysial connective tissue and focal fatty infiltration (a), rounded myofibers and internal nuclei (b). Normal checkerboard pattern is lost and oxidative (type I) fibers are predominant in SDH stain (c). Few regenerating fibers are present by neonatal myosin stain (d). (All original magnification × 20).
At nerve conduction studies sural sensory nerve action potential amplitudes were below the normal limits (<6.5 μV), which suggests a mild axonal sensory neuropathy. Sensory evoked potentials from median and posterior tibial nerves revealed central conduction time (CTT) prolongation and indicated bilateral involvement of dorsal column-medial lemniscal sensory pathways (Supplementary Figure 2). CTT for posterior tibial nerve (N20-P40 interpeak latency) was 22.7 ms at right side and 22.1 ms at left side (laboratory upper limit is 21.0 ms); while for median nerve (N13-N20 interpeak latency) it was 9.3 ms at right side, and 10.2 ms at left side (laboratory upper limit is 7.2 ms). Furthermore, the amplitudes of the spinal potentials were low.
Due to the genetic heterogeneity of distal AMC, Mendeliome sequencing was used to uncover the genetic cause after informed consent. Genomic DNA of the index patient was enriched for 4813 genes associated with known clinical phenotypes (from OMIM, ‘mendeliome’ panel) with the True Sight One Panel (Illumina, San Diego, CA, USA) and sequenced on an Illumina High Seq 4000 Sequencer as described previously.5
Variants were filtered for a recessive inheritance model with consanguine familiar background. Four variants fitted the filter criteria (Supplementary Table 1). Three were known variants that did not fit the phenotype; the fourth variant was an undescribed stop mutation in piezo-type mechanosensitive ion channel component 2 (PIEZO2). The C to T exchange at position 7712 in the cDNA leads to the introduction of a premature stop codon terminating the protein at amino-acid position 462 (NM_022068, c.1384C>T p.R462*, Figure 3).
Figure 3
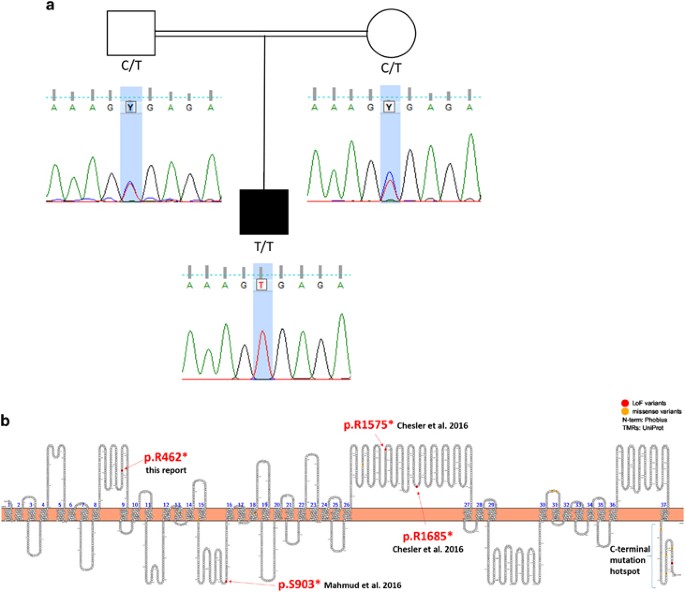
Mutation analysis. (a) Pedigree including the sequence chromatograms of the family showing the autosomal recessive inheritance. Consanguineous parents are heterozygous whereas the affected offspring carries the homozygous stop mutation (NM_022068, c.1384C>T p.R462*). (b) Schematic representation of PIEZO2 transmembrane topological structure based on the prediction from UniProt plotted with Protter (http://wlab.ethz.ch/protter). The locations of reported LoF mutations in this report and in the publications by Mahmud et al. and Chesler et al. are highlighted in read, and indicated with arrows. Piezo proteins likely form tetramers18 or trimers19 to build the active channels. This multimerization could explain the dominant effect observed in distal AMC. Disease causing mutations described for PIEZO1 and PIEZO2 cluster at the c-terminal end of the proteins.7 Coste et al.6 observed increased channel activity in HEK cells transfected with human PIEZO2 plasmids carrying two different mutations and concluded a gain of function in their patients. As the c-terminal end of the protein likely forms the ion-conducting pore, mutations in this domain may well lead to altered channel activity or sensitivity. The here-described stop mutation locates to the forth cytoplasmic loop of the predicted transmembrane structure of PIEZO2 and is predicted to lead to NMD (by Mutation Taster) and likely results in complete loss of PIEZO2 protein or alternatively in the production of a severely truncated protein.
Heterozygous mutations in PIEZO2 have been described in autosomal-dominant distal AMC and related diseases.6, 7, 8 Piezo proteins are highly conserved nonselective cation channels that are expressed in multiple mechanosensitive tissues9 such as, spinal sensory neurons, endothelium and visceral tissues. Piezo1 has a critical role in vascular remodeling and red blood cell volume regulation,10, 11, 12 while Piezo2 is a mechanotransducer for low threshold proprioception in murine skin.13
Proprioception is the perception of body and limb position, and is mediated by proprioceptors which are sensory neurons with cell bodies in the dorsal root ganglia and innervate two mechanoreceptors in skeletal muscles: muscle spindles and Golgi tendon organs.13
Complete knockout of Piezo2 in mouse is perinatal lethal.14 Mice that lack Piezo2 in mechanosensory neurons show severely impaired limb coordination, stiff limbs and unstable gait.13 The proprioceptive neurons in tissue-specific knockout animals were similar in number and sensory endings to those from wild-type animals but were defective in mechanically induced action potential firing.13
Besides proprioception PIEZO2 may also have other functions, it is expressed in epithelial cells and upregulated during muscle inflammation and tumor angiogenesis.15, 16 Interesting in this context is the myopathy confirmed by muscle biopsy in the patient with regenerating fibers. Low level of PIEZO2 expression in muscle has been shown in several online databases and mechanotransduction via PIEZO2 seem to be essential for muscle hemostasis. Detailed description and appreciation of myopathy in genetically defined Marden–Walker syndrome cases has been lacking so far.17 The underlying mechanism leading to myopathy due to PIEZO2 mutations needs to be elucidated by future research. The here-described mutation is predicted to have a loss-of-function pathogenesis, whereas the dominant mutations on the c-terminal hotspot may act dominant-negatively on the wild-type allele because PIEZO2 likely forms multimers.18, 19 Compared with dominant phenotypes,6, 7, 8 our patient despite having dysmorphic facial features did not have deep-set eyes, micrognathia, cleft palate or a bifid uvula, ptosis, camptodactly, blepharophimosis and short stature. The clinical phenotype was compatible with distal arthrogryposis type 5, in terms of progressive pulmonary insufficiency because of restrictive chest due to scoliosis, and worsening of contractures.
However, distinguishing characteristics of the patient have been loss-of-joint position sense, mild sensory neuropathy, incoordination and unsteadiness due to posterior column involvement, which were also demonstrated electro physiologically. Although this report was under review, two independent groups published additional cases with homozygous or compound heterozygous loss-of-function mutations in PIEZO2.17, 20 The patients presented in these publications show a similar phenotype to our patient with defects in proprioception but myopathy had not been well appreciated. It seems that loss-of-function mutations in PIEZO2, albeit rare, comprise an under diagnosed disease entity.
In summary, we present a clinically well-described patient with almost 15 years follow-up data harboring a homozygous stop mutation in PIEZO2 leading to distal AMC. The observed sensory ataxia and proprioceptive defect show marked similarity to the knockout mice models for piezo2. Sensory ataxia and proprioception defect with dorsal column involvement together with arthrogryposis, scoliosis, and slow progressive respiratory deterioration may represent a distinct clinical phenotype, and may indicate mutations in PIEZO2.