Characterization of the 1918 influenza virus polymerase genes (original) (raw)
Influenza A viruses cause annual outbreaks in humans and domestic animals. Periodically, new strains emerge in humans that cause global pandemics. The severe ‘Spanish’ influenza pandemic of 1918–1919 infected hundreds of millions, and resulted in the death of approximately 50 million people12. We have previously used phylogenetic analyses to help understand the origin of the pandemic virus8,11; functional studies to understand the pathogenicity of the 1918 virus are underway6,13,14,15,16,17. Recent data have shown that viral constructs bearing the 1918 haemagglutinin gene are pathogenic in a mouse model, but the genetic basis of this observation has not yet been mapped6,13,14,15,16,17. The overall goals of this project have been to understand the origin and unusual virulence of the 1918 influenza virus.
The influenza virus A polymerase functions as a heterotrimer formed by the PB2, PB1 and PA proteins (see ref. 1 for a review). An additional small open reading frame has recently been identified, coding for a peptide (PB1-F2) that is thought to play a role in virus-induced cell death18. It is not yet clear how the polymerase complex must change to adapt to a new host3. A single amino acid change in PB2, E627K, was shown (1) to be important for mammalian adaptation2,3, (2) to distinguish highly pathogenic avian influenza (HPAI) H5N1 viruses in mice19, and (3) to be present in the single fatal human infection during the HPAI H7N7 outbreak in the Netherlands in 2003 (ref. 20), and in some recent H5N1 isolates from humans in Vietnam and Thailand and wild birds in China21,22,23.
The open reading frame sequences of segment 1 (PB2), segment 2 (PB1) and segment 3 (PA) of A/Brevig Mission/1/1918, and theoretical translations of the four identified reading frames, are shown in Supplementary Fig. 1a–c. The 1918 PB2 protein contained five changes from the avian consensus sequence (Table 1). Of these, A199S is in the area mapped as the PB1 binding site, and the L475M change is in a nuclear localization signal24,25,26. Three other changes at residues 567, 627 and 702 occur at sites that are not in known functional domains.
Table 1 Amino acid residues distinguishing human and avian influenza polymerases
The 1918 PB1 protein differed from the avian consensus by seven residues (one of which is shown in Table 1; see also Supplementary Fig. 2). Of these, K54R is in the overlapping binding domains for complementary (c)RNA and viral (v)RNA. Changes at residues 375, 383 and 473 all occur in between the four conserved polymerase motifs in the cRNA binding domain27, and changes at residues 576, 645 and 654 occur in the vRNA binding domain28.
Seven changes were noted in the 1918 PA protein compared with the avian consensus (four of which are shown in Table 1, the other three being C241Y, K312R and I322V). The C241Y change occurs in a nuclear localization signal, but the other six changes (at residues 55, 100, 312, 322, 382 and 552) occur at sites outside of known functional domains24,25,26.
Representative phylogenetic analyses of the three polymerase genes are shown in Figs 1–3. The 1918 human pandemic viral polymerase genes were compared to representative avian influenza genes with regards to transition/transversion (T i/T v) ratio, synonymous/non-synonymous (S/N) ratio, and the numbers of differences at fourfold degenerate sites (defined in ref. 11). T i/T v ratios for most comparisons using the 1918 viral genes and representative sequences of either North American or Eurasian avian genes yielded values between 2 and 4. This range was similar to that observed for comparisons of various avian genes with one another, except for the PB1 gene. For PB1, comparisons of the 1918 viral gene with avian virus PB1 genes was always close to 2, whereas comparisons of various avian genes with one another were in the range of 6–10. There were fewer transversions in comparisons between avian PB1 genes than in comparisons between avian PB1 and 1918 human virus PB1, probably reflecting that transversions more often lead to non-synonymous changes.
Figure 1: Phylogenetic tree of the PB2 gene.

Sequences were aligned and analysed for phylogenetic relationships using the NJ algorithm, with the proportion of sequence differences as the distance measure. Bootstrap values (100 replications) for key nodes are shown (for clarity, identical and nearly identical sequences have been removed from the trees). Major clades are identified with large brackets. The arrow identifies the position of the 1918 PB2 gene sequence. A distance bar is shown below the tree. Influenza strain abbreviations used in the analyses are listed in Supplementary Table 1.
Figure 2: Phylogenetic tree of the PB1 gene.
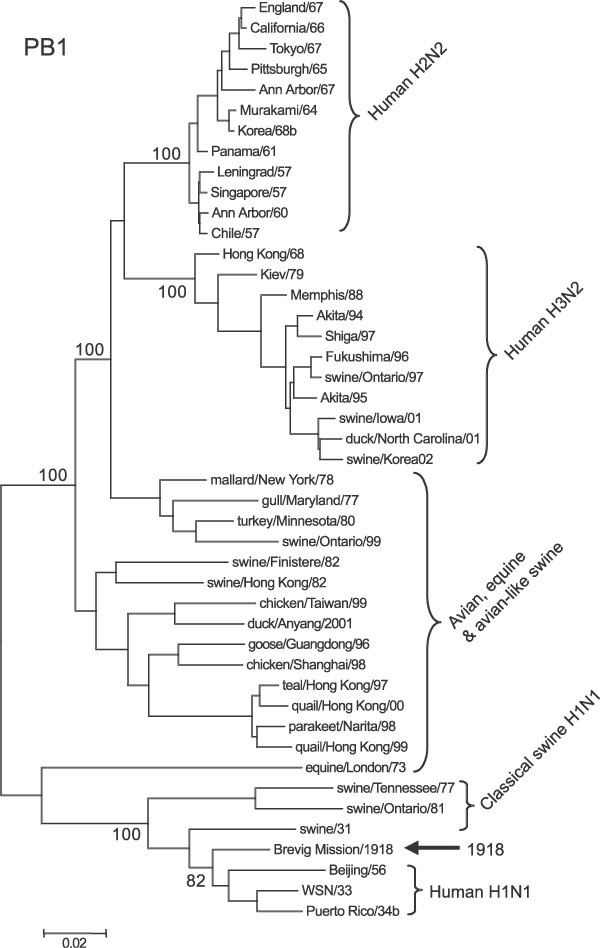
Sequences were aligned and analysed as detailed in the legend to Fig. 1.
Figure 3: Phylogenetic tree of the PA gene.
Sequences were aligned and analysed as detailed in the legend to Fig. 1.
S/N ratios for most comparisons using the 1918 viral genes and representative sequences of either North American or Eurasian avian genes usually yielded values in the range of 7–16 for both the PA and PB2 genes, as is the case for most avian versus avian PA and PB2 gene comparisons. Like the T i/T v ratios, the S/N ratios were somewhat higher with the PB1 gene (most of the comparisons yielded ratios in the range of 16–25), owing to a smaller number of non-synonymous changes in comparisons of avian PB1 genes with one another. These findings may reflect a more conservative evolution of PB1 in birds.
A subset of synonymous differences occurs at sites that are fourfold degenerate (that is, where a substitution with any base does not result in an amino acid replacement). As these sites are not subject to selective pressure at the protein level, base substitutions at many fourfold degenerate sites may accumulate rapidly. If influenza virus genes have been evolving in birds for long enough to reach evolutionary stasis, as is suggested by the high S/N ratios described above, one would predict that at many of the sites where fourfold degeneracy is possible, all four bases would be present in the avian clade unless the constraints of RNA secondary structure limit the accumulation of synonymous changes. In fact, when avian sequences from geographically distinct lineages (North American versus European) were compared, the per cent difference at fourfold degenerate sites yielded values in the 27–38% range. In contrast, calculating the per cent difference at fourfold degenerate sites in comparisons of the 1918 viral PA, PB1 and PB2 gene sequences with avian sequences yielded consistently higher values (range 41–51%) for all three genes. As with the other 1918 genes11, this suggests that the donor source of the 1918 virus was in evolutionary isolation from those avian influenza viruses currently represented in the databases.
Emphasizing the avian-like nature of the 1918 influenza virus polymerase proteins, out of 19 total amino acid changes from the avian consensus, there are only 10 amino acid positions (out of 2,232 total codons) that consistently distinguish the 1918 and subsequent human polymerase proteins PB2, PB1 and PA from their avian influenza counterparts (these are defined as changes from avian sequences in the 1918 virus that are maintained without change in subsequent human viruses) (Table 1). It is likely that these changes have an important role in human adaptation. Seven of these ten changes were previously noted in an alignment between avian and human influenza polymerases3. What follows is a comparison between the 1918 virus changes and recent H5N1 isolates, in order to evaluate possible examples of parallel evolution in the adaptation of avian influenza viruses to humans.
In the PB2 protein, five changes distinguish the human isolates from avian sequences (Table 1). Out of 253 available PB2 sequences from human H1N1, H2N2 and H3N2 isolates, these five changes are almost completely preserved, with the exception that two recent H3N2 isolates have the avian Lys residue at position 702. Only a small number of avian influenza isolates show any of these five changes, and it is intriguing that almost all of these isolates are from HPAI H5N1 or H7N7 viruses, or from the H9N2 lineage that infected a small number of humans in China in the late 1990s (ref. 29). Only 5 out of 282 available avian PB2 sequences have a Ser residue at position 199, four of these being 1997 H5N1 isolates from Hong Kong. The A199S change was also found in 5 out of 18 H5N1 strains isolated from humans (all five were from the 1997 Hong Kong outbreak). Of the avian viruses, 36 out of 336 have an Arg residue at position 702, 30 of which are H9N2 isolates from China around 1996–2000, and 5 are H5N1 isolates from Hong Kong in 1997 and 2001. Out of 18 available 1997 H5N1 strains isolated from humans, three have the K702R change.
Perhaps most interestingly, the 1918 virus and subsequent human isolates have a Lys residue at position 627. This residue has been implicated in host adaptation2,3, and has previously been shown to be crucial for high pathogenicity in mice infected with the 1997 H5N1 virus19. Of the avian isolates, 19 out of 345 have a Lys residue at position 627, 18 of which are HPAI H5N1 or H7N7 avian influenza viruses. Sixteen of these were recently characterized H5N1 isolates from a die-off of wild waterfowl around Qinghai Lake in western China in 2005 (ref. 21). In human H5N1 isolates, 11 out of 37 have the E627K change: A/Hong Kong/483/1997 and A/Hong Kong/485/1997, four out of six isolates from Vietnam in 2004 (ref. 22), and two out of three isolates from Thailand in 2004 (ref. 23). The E627K mutation was seen in six out of seven H5N1 isolates from Thai tigers in 2004, and was also present in the H7N7 virus responsible for the single human fatality during the HPAI H7N7 outbreak in the Netherlands in 2003 (ref. 20). It was not noted in the contemporaneous chicken isolates.
At position 475, only one out of 355 avian isolates has a Met residue (an H5N1 HPAI virus from 2004). Similarly, only one out of 345 avian viruses has an Asn residue at position 567. None of the human H5N1 isolates has the L475M or the D567N changes. None of the available H5N1 or H7N7 sequences has more than one of the proposed human-adaptive PB2 changes determined for the 1918 virus.
The PA protein shows a similar pattern: four residues consistently differ between 1918 and subsequent human isolates and the avian consensus sequence (Table 1). Three other changes (C241Y, K312R and I322V) distinguish 1918, H1N1 and H2N2 human isolates, but most H3N2 isolates have the avian amino acid at these positions. Of 295 available sequences from human H1N1, H2N2 and H3N2 isolates, all have Asn at position 55 (except A/WSN/33), Ala at position 100 and Ser at position 552. Only 5 out of 295 human isolates have the avian Glu residue at position 382. Notably, these five isolates make up a minor clade of recent H3N2 isolates that have a number of unusual changes from typical human H3N2 viruses30. When avian influenza sequences are analysed, none (out of 209 sequences) has Asn at position 55 or Ser at position 552. Only 8 out of 209 avian PA protein sequences show the V100A change: six recent H6N2 isolates from chickens in California, and two HPAI 2002 H5N1 duck isolates from China. Of the 209 avian sequences, five have an Asp residue at position 382, including two HPAI H5N2 isolates from chickens in Mexico in 1994.
The PB1 gene segment was replaced by reassortment in both the 1957 and 1968 pandemics9. We compared the PB1 protein from the 1918 human virus with those of the avian-derived PB1 segments from the 1957 and 1968 pandemics. Human H1N1, H2N2 and H3N2 viruses derived from the 1918, 1957 and 1968 pandemics, respectively, each possessed a uniquely derived avian-like PB1 gene segment, and so we sought to identify any parallel changes that might shed light on human adaptation. The three human pandemic PB1 proteins differ from the avian consensus by only 4–7 residues each (Supplementary Fig. 2). Only one of these changes is shared among the pandemic isolates: an N375S change. This change to a serine residue is also found in swine and equine influenza A isolates. With few exceptions, all human influenza PB1 proteins have Ser at this site. Of 230 human influenza sequences, only two H1N1 isolates (A/FM/47 and A/Beijing/1956) and the ‘minor clade’ H3N2 isolates described above have the avian Asn residue30. In contrast, although this residue is maintained in almost all mammalian isolates, it is variable among avian PB1 proteins. Of 293 avian isolates, 66% have the consensus Asn residue at position 375, 18% have a Ser residue and 12% have a Thr residue.
The data presented here highlight the marked conservation of the PB1 protein in avian influenza viruses. PB1 functions as an RNA-dependent RNA polymerase, and so it is reasonable to hypothesize that its enzymatic function is optimal in this conserved form. In humans, the PB1 proteins experience linear change over time. Indeed, PB1 in humans acquires ∼0.4 amino acid changes per year. As there is such strong antigenic selection on human viruses, it is possible that although the observed changes in PB1 are selectively beneficial with respect to antigenicity, they are mildly deleterious to enzyme function. Such complex fitness trade-offs are thought to be commonplace in RNA virus evolution. Supporting this hypothesis, a recent study examining combinations of avian and human influenza polymerases showed that the most efficient influenza transcriptional activity in vitro was seen with an avian-derived PB1, even if the PB2, PA and NP proteins were from a human virus3. Acquiring an avian PB1 by reassortment might provide a replicative advantage to the new virus, possibly explaining why both of the last two pandemics and the 1918 influenza virus all had very avian-like PB1 proteins.
Both the 1957 and 1968 pandemic influenza viruses were avian/human reassortants in which 2–3 avian gene segments were reassorted with the then-circulating, human-adapted virus9,10. Unlike the 1957 and 1968 pandemics, however, the 1918 virus was most likely not a human/avian reassortant virus, but rather an avian-like virus that adapted to humans in toto8,11. On the basis of amino acid replacement rates in human influenza virus polymerase genes, it is possible that these segments were circulating in human influenza viruses as early as 1900. However, proof that the 1918 virus did not retain gene segments from the previously circulating human influenza A strain would require discovery of a sample of the pre-1918 virus from archival material. The donor source, although avian-like at the protein level, may have come from a subset of avian influenza viruses not currently represented in the sequence databases and may have been in evolutionary isolation.
The fact that amino acid changes identified in the 1918 analysis are also seen in HPAI strains of H5N1 and H7N7 avian viruses that have caused fatalities in humans is intriguing, and suggests that these changes may facilitate virus replication in human cells and increase pathogenicity. It is possible that the high pathogencity of the 1918 virus was related to its emergence as a human-adapted avian influenza virus. These changes may reflect a process of parallel evolution as avian influenza A viruses mutate in response to adaptational pressures, and suggest that the genetic basis of avian influenza virus adaptation to humans can be mapped.