Mapping determinants of human gene expression by regional and genome-wide association (original) (raw)
The expression level of genes, ‘the expression phenotype’3, is highly variable and heritable in humans4,5 and other organisms6. We previously1 performed genome-wide linkage analysis for 3,554 expression phenotypes in 14 pedigrees from the Utah component of the Centre d'Etude du Polymorphisme Humain (CEPH) to map the genetic determinants of variation in human gene expression. For ∼1,000 expression phenotypes, significant linkage evidence was obtained, suggesting the existence of determinants that act in cis or trans to the expressed gene. In the present study, we used genotype data generated by the International HapMap Project2 to test for allelic association in two sets of analyses. First, for a set of 374 phenotypes with evidence of _cis_-linked determinants, we performed association analysis with dense sets of SNPs near linkage peaks. Second, we restricted attention to the 27 phenotypes with the strongest linkage evidence for _cis_-acting determinants, and tested >770,000 SNPs in the genome for association. Because all the phenotypes have been mapped previously by genetic linkage, the analyses allow us to compare directly the results from whole-genome linkage and association studies.
The 374 phenotypes have at least one marker with linkage evidence (t > 2) for cis regulators1; this corresponds to a point-wise P < 0.02 with the sample of 14 CEPH sibships. For the present analysis, we obtained SNP genotype data on 57 unrelated CEPH individuals from the International HapMap Project2 and generated expression phenotypes using the Affymetrix Human Genome Focus arrays.
Evidence for linkage requires co-segregation between the phenotype and a marker site, but does not depend on the particular allele present at the marker. In contrast, allelic association with a linked marker requires correlation with a particular SNP allele; that is, linkage disequilibrium. Even if there are several different alleles at the determinant (‘allelic heterogeneity’), linkage can be detected. But if there is allelic heterogeneity, it is less likely that there will be detectable association. Therefore, it was not obvious that evidence for linkage would predict evidence for association. So, for a set of phenotypes with cis linkage, we performed association analysis with SNPs within the target genes and within 50 kilobases (kb) of the 5′ and 3′ ends, and compared results with those from the previous linkage scans1. The evidence for association was assessed by linear regression. Among the 374 phenotypes, there are 65 (17%) with at least one marker that shows evidence of association at the nominal P < 0.001 level. For some of the phenotypes, the association with a nearby marker is extremely strong; among the 65 phenotypes, there are 12 with evidence of _cis_ association at _P_ < 10-10. At the less stringent threshold of _P_ < 0.01 for association, there are 133 (36%) phenotypes. We also determined the proportion of phenotypes with these two nominal levels of evidence for _cis_ association for various strengths of initial linkage findings (Supplementary Table 1). We found that the strength of linkage evidence did tend to predict association results. For example, among the 27 phenotypes with highly significant _cis_ linkage (_t_ > 5, P < 3.7 × 10-5), 70% have evidence of cis association at P < 0.001, compared to only 9% of the phenotypes with modest evidence of cis linkage (2 < t < 3, P < 0.02).
Although there are many examples of regulatory sites located in 5′ or 3′ flanking regions of genes, little is known about the relative frequencies. Although the marker most strongly associated with gene expression level is not necessarily the functional variant, we expect that in most cases that marker will be very close to the functional variant. With this assumption, we determined the location of the markers within 50 kb of the target genes that showed the strongest association, to establish whether they occur preferentially in the 5′ or 3′ regions. Among the 133 phenotypes with cis association at P < 0.01, the regulatory sites are found in approximately the same proportions in the 5′ (27%) and 3′ (34%) ends, and within the target genes (25%). For 14% of the phenotypes, linkage disequilibrium among SNPs spanning the regions examined was so strong that we were not able to narrow the regions of cis association. Thus, overall we found that cis regulators are not preferentially enriched in the 5′ or 3′ regions around the target genes. However, for most of the phenotypes, the analysis of regional association data narrowed the search for the regulatory determinants to one particular region near the target gene.
The analyses described so far were restricted to the SNPs known to be located in regions near the target genes. If we did not know in advance where to look for determinants, how successfully would we find them? To answer this question, we took advantage of the hundreds of thousands of markers across the genome, genotyped on the same 57 unrelated CEPH individuals as above. Instead of focusing on cis regulatory regions, we performed genome-wide association analysis (GWA) to map determinants. We limited our analysis to the 27 phenotypes with the strongest evidence of cis regulation from our whole-genome linkage analysis1 so that the results could be compared with the linkage results in which we have highest confidence. We tested 770,394 SNP markers for association with each expression phenotype and performed a regression analysis of expression level on marker genotype. We used the Šidák procedure, which is conservative7, to correct for multiple testing.
The evidence for association was significant at the genome-wide level (nominal P < 6.7 × 10-8 or corrected P < 0.05) for 14 of the 27 phenotypes (Table 1). Figure 1 shows several examples. The GWA identified only cis regulation for 12 of the phenotypes, cis and trans regulation for another phenotype (phosphoribosyl pyrophosphate aminotransferase (PPAT)), and only trans regulation for one phenotype (DEAD box polypeptide 17 (DDX17)).
Table 1 Genome-wide association results for 27 phenotypes
Figure 1: Results of genome-wide association analysis for six representative phenotypes with cis regulators.
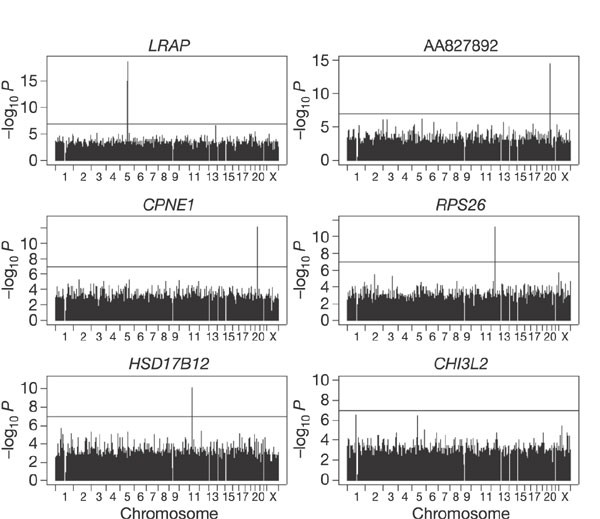
The horizontal line in each panel corresponds to P = 0.05 after Šidák correction.
We compared the findings for the 27 phenotypes in Table 1 to those from our previous whole-genome linkage scans1. To simplify this analysis, we focused on the SNP with the most significant finding in the GWA for each phenotype in Table 1. For this SNP, we classified the results according to whether the association was or was not supported by the genome-wide linkage analysis (that is, whether the SNP that showed significant association fell within the region of significant linkage). For 15 of the 27 phenotypes, genome-wide linkage analyses and GWA pointed to the same cis regulatory regions. In 13 of these, the corrected _P_-value for the cis marker was <0.05 (_P_uncorr. < 6.7 × 10-8) in the GWA, providing highly significant evidence for cis regulatory elements. In one of the 13 phenotypes, PPAT, the GWA result points to both cis and trans regulators; however, linkage scan results support only the cis regulation. For the other two phenotypes (POMZP3 and CHI3L2) the results were nominally significant (P = 7 × 10-8 and P = 3 × 10-7, respectively). For both phenotypes, the peak SNPs are located close to the target genes (6 kb 5′ for POMZP3 and 91 base pairs (bp) 5′ for CHI3L2). Because of the location, we consider these cases ‘true’ positives, but recognize that the statistical evidence alone is not compelling. We show evidence below that one of these peak SNPs is a regulatory variant of CHI3L2.
For the remaining 12 of the 27 phenotypes, no strong evidence for cis determinants was found by GWA. In 11 of the 12 phenotypes, no significant association (_P_corr. < 0.05 or _P_uncorr. < 6.7 × 10-8) was detected anywhere in the genome, even though highly significant evidence for cis regulation was detected in the linkage scans. For DDX17, as indicated above, GWA identified significant trans association (_P_corr. < 0.05) but evidence for a _trans_-acting regulator was not found in the linkage scan. For all 12 phenotypes, the most significant markers were located on chromosomes different from that of the expressed gene. (There are 13 ‘_trans_’ markers listed in Table 1; one is the non-cis marker for PPAT, see above.) The locations and _P_-values from the association analyses for these markers are shown in Table 1. In some cases, the failure to find significant evidence for cis association, despite highly significant linkage results for cis regulation, may be due to linkage findings that were false-positive results. However, even if the linkage findings reflect real effects, allelic heterogeneity might make the determinant undetectable by association. Furthermore, our sample size of 57 might be too small to yield statistically significant associations, especially if the marker closest to a determinant is not in strong linkage disequilibrium with it.
To assess the issue of sample size for this and future studies, we determined the power to detect association—with the same correction we used for multiple testing—under various assumptions about sample size. We also considered variation in effect size; that is, the proportion (_R_2) of phenotype variation accounted for by the SNP. As a point of reference, the observed values of _R_2 for the 13 phenotypes with significant cis association range from 0.44 to 0.78. (In classical complex traits and diseases, _R_2 for individual determinants is expected to be much smaller.) To achieve a probability of 0.8 of detecting a determinant for which _R_2 is 0.1, we found that samples of ∼500 would be needed (see Supplementary Information). Even these results must be viewed as optimistic; the calculations apply to an idealized determinant that is identical to the marker in both location and frequency, yielding maximum linkage disequilibrium. Furthermore, it is difficult to extrapolate the results to complex traits in general. For many quantitative traits of interest, the fraction of phenotypic variance attributable to any one determinant is likely to be far smaller than the values we estimated for the 13 strongly associated _cis_-linked expression phenotypes. Thus, extrapolations from the small samples used here to genome-wide analysis with other complex traits should be done cautiously—much larger samples will usually be required.
We expected that the much greater resolution afforded by linkage disequilibrium would result in candidate regions defined by association that are much smaller than those defined by linkage. For all 15 phenotypes where the linkage and GWA results are concordant, this expectation was confirmed. Figure 2 shows several examples. For example, in the GWA for CPNE1, the 30 markers with significant evidence of association (_P_corr. < 0.05) span a region of only 402 kb. For IRF5, the ten significant markers span 90 kb of chromosome 7. For four phenotypes (RPS26, GSTM1, GSTM2 and DDX17), there is only one significant marker.
Figure 2: Results of genome-wide linkage analysis (dotted line) superimposed on those from genome-wide association (bars) for the chromosome where the expressed gene is located.
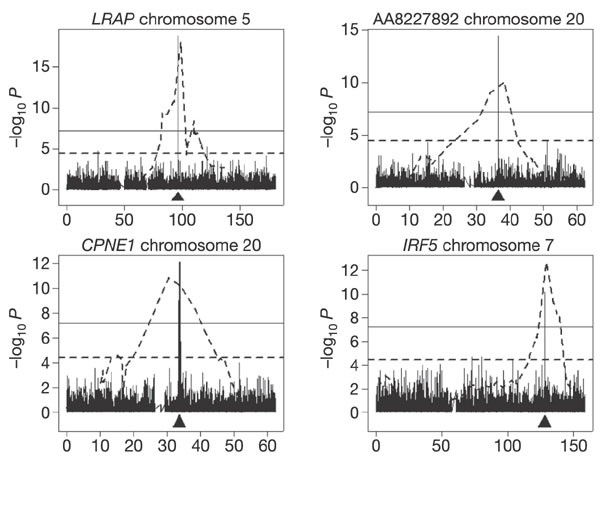
The location of the expressed gene is indicated by an arrow. The dotted horizontal line is for data from linkage scans and corresponds to t = 4, P = 3.7 × 10-5. The solid horizontal line is for data from GWA and corresponds to P = 0.05 after Šidák correction. The x axis indicates chromosome location in megabases.
Ultimately, the findings from linkage and association need to be confirmed by functional analysis. Results from our genome-wide linkage and association analyses suggest that expression level of chitinase 3-like 2 (CHI3L2) is regulated by a _cis_-acting transcriptional regulator (Table 1). A marker (rs755467) in the promoter region of CHI3L2 shows nominally significant association results (P < 3 × 10-7). Although it is not significant after correction for 770,394 markers, we considered it likely that expression level of _CHI3L2_ is indeed regulated by a polymorphism in its promoter region, because both linkage and association results point to the same candidate regulatory region. We followed up this finding with functional tests. To evaluate the relative promoter activity of the haplotypes bearing the different alleles of the SNP marker rs755467, we performed luciferase reporter assays. Stronger luciferase expression (> 3-fold) was observed for vectors containing the upstream fragment than for the empty vector, confirming promoter activity in the DNA fragment containing marker rs755467. We then compared the promoter activity of the fragment bearing the rs755467 T-allele with that bearing the G-allele. The promoter activity of the fragment bearing the rs755467 T-allele was 2 ± 0.6-fold higher (P < 0.05) than that bearing the G-allele.
Next, we performed haplotype-specific chromatin immunoprecipitation (haploChIP) to determine whether this observation is due to differential binding of RNA polymerase II to the different haplotypes8,9. The assay was performed using lymphoblastoid cells from three CEPH individuals heterozygous at rs755467. We used antibody against RNA polymerase II (phosphorylated serine 5), which is known to accumulate in promoter regions of genes10. We quantified the relative abundance of the RNA polymerase II binding to the haplotypes bearing the T- or G-alleles in each of the heterozygous individuals by quantitative polymerase chain reaction (PCR)9. In all three cases, we found that in immunoprecipitated DNA, the amount of DNA bearing the T-allele of rs755467 is much higher than that bearing the G-allele (≥ 90% T and 10% G) (Fig. 3). This result suggests that the higher expression of CHI3L2 in individuals who are TT homozygotes for rs755467 is due to stronger binding of RNA polymerase II to DNA containing the T-allele than to that containing the G-allele. Thus, results from both functional assays support data from the association analysis and suggest that the polymorphism rs755467 or a marker in strong linkage disequilibrium with it is a determinant that affects the expression level of CHI3L2.
Figure 3: HaploChIP assay with RNA polymerase II antibody to estimate allele-specific binding.
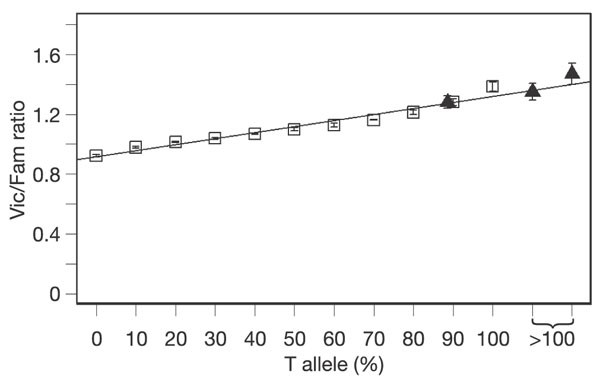
Open squares show fluorescence intensities (Vic/Fam ratios) of genomic DNA samples with experimentally varied proportions of allele T at SNP rs755467. The linear regression line is shown. Solid triangles show mean fluorescence intensities of immunoprecipitated products from three individuals heterozygous at rs755467. In all three cases, the immunoprecipitated products contained more T than G in the DNA. Estimates greater than 100% for the T-allele imply that no G-allele-bearing DNA fragments were detected in the immunoprecipitated products. Standard deviation of triplicate measurements is shown.
The results of this study have implications for studies of many phenotypes besides gene expression. It has become a truism that finding genes that contribute to complex traits and diseases is much more difficult than finding genes for mendelian traits, and the underlying problems have been discussed in many reviews11,12,13,14. As genomic resources have become available, increasing attention has been given to the role of association analysis, first described systematically in ref. 15. Here we have carried out association analysis with dense SNP maps in two settings that parallel approaches used to map genes for human diseases and traits. First, we performed regional association analysis with SNP markers in and around target genes to identify _cis_-acting regulatory variants. This approach parallels candidate gene studies. Second, we analysed >770,000 SNP markers to identify determinants of variation in gene expression by GWA analysis. This parallels studies where no candidate regions are specified in advance, and association analysis is used to find susceptibility genes. The GWA analysis, carried out with only 57 unrelated individuals, yielded highly significant results for 14 of the 27 phenotypes. The evidence for association was so strong in these cases that the findings remained significant (_P_corr. < 0.05) even after correction for the large number of tests genome-wide.
The use of GWA to find genes is controversial12,16,17. The optimism inspired by the availability of very dense SNP maps has been tempered by the resulting problems of multiple testing. There have been a few genome-wide association studies18,19,20; however, the marker density in those studies was much lower than that made possible here by the International HapMap Project. Despite the small sample size, we obtained significant findings for approximately 50% of the 27 phenotypes analysed by genome-wide association analysis. The results suggest that as denser marker maps become available and high-throughput genotyping technologies are developed, large-scale association studies will become a practical means to identify genes for complex traits and diseases.