Rethinking schizophrenia (original) (raw)
- Perspective
- Published: 10 November 2010
Nature volume 468, pages 187–193 (2010)Cite this article
- 126k Accesses
- 57 Altmetric
- Metrics details
Subjects
Abstract
How will we view schizophrenia in 2030? Schizophrenia today is a chronic, frequently disabling mental disorder that affects about one per cent of the world’s population. After a century of studying schizophrenia, the cause of the disorder remains unknown. Treatments, especially pharmacological treatments, have been in wide use for nearly half a century, yet there is little evidence that these treatments have substantially improved outcomes for most people with schizophrenia. These current unsatisfactory outcomes may change as we approach schizophrenia as a neurodevelopmental disorder with psychosis as a late, potentially preventable stage of the illness. This ‘rethinking’ of schizophrenia as a neurodevelopmental disorder, which is profoundly different from the way we have seen this illness for the past century, yields new hope for prevention and cure over the next two decades.
Similar content being viewed by others
Main
The challenge of creating a vision of schizophrenia for 2030, which I attempt here, is a difficult one. There is certainly a risk in predicting scientific progress—the most important discoveries will probably be ones we cannot imagine today. But it is equally true that we can use past experience and the present state of knowledge to predict some aspects of the future. For schizophrenia, our knowledge base in 2010 is mostly based on clinical observation.
Schizophrenia is a syndrome: a collection of signs and symptoms of unknown aetiology, predominantly defined by observed signs of psychosis. In its most common form, schizophrenia presents with paranoid delusions and auditory hallucinations late in adolescence or in early adulthood. These manifestations of the disorder have changed little over the past century.
A century ago we had large public institutions for serious mental illness, tuberculosis and leprosy. Of these three, today only mental illness, especially schizophrenia, remains unchanged in prevalence and disability1.
Sustained recovery occurs in less than 14% within the first five years following a psychotic episode2. Longer-term outcomes may be marginally better: a large international 25-year follow-up study reported an additional 16% with late-phase recovery3. Throughout Europe, less than 20% of people with schizophrenia are employed4. A large US study found nearly 20% homeless in a one-year follow up5. And a recent report from a patient advocacy group reported that in the US those with serious mental illness were three times more likely to be found in the criminal justice system than in hospitals. (http://www.treatmentadvocacycenter.org)
Although many have attributed this lack of progress to failed systems of care (http://www.mentalhealthcommission.gov/), we still do not have a basic understanding of the pathophysiology of the disorder and therefore lack the tools for curative treatment or prevention needed for most people with schizophrenia. If we are to transform outcomes by 2030, we can start by offering individuals and families challenged by serious mental illness a candid account of the current state of knowledge and a thoughtful consideration of future prospects.
One-hundred years of schizophrenia
The history of schizophrenia says more in many ways about the perspectives of the observer than the observed. In the late nineteenth century, Kraepelin defined “dementia praecox” or premature dementia as distinct from the insanity of tertiary syphilis or the cyclic, non-deteriorating psychosis of manic depressive illness6. Bleuler, who coined the term schizophrenia in the early twentieth century, was less convinced of its deteriorating course but emphasized the notion of a fundamental disorder of thought and feeling, which every psychiatrist for decades learned as the four ‘a’s—disturbances of associations, affect, ambivalence and autistic isolation7. These early formulations emerging before the split between neurology and psychiatry were consistent with the notion of a mental disorder rooted in brain pathology. Yet for much of the twentieth century, with the predominance of psychoanalytic theory, the study of the mind ignored the brain. Schizophrenia was a psychotic reaction, a fragmented ego due to a rejecting or ambivalent mother and treatments included re-mothering to build a stable ego8.
In the second half of the twentieth century, with the emergence of neuroleptic drugs, the pendulum swung in the other direction—a focus on brain chemistry deemphasized the mind. Schizophrenia was considered a ‘dopamine disorder’ based on the psychosis-inducing effects of dopamine-releasing drugs, such as amphetamine, and the anti-psychotic efficacy of a score of drugs that blocked the dopamine D2 receptor9. This neurochemical view of schizophrenia yielded medications that transformed the treatment of psychosis, allowing patients to be treated outside of hospitals and, in some cases, resulting in remission of the major symptoms of the illness. Early neuroleptic medications, examples of which are chlorpromazine and haloperidol, have been increasingly replaced by ‘atypical’ antipsychotics that have fewer extrapyramidal side effects (such as tremor and rigidity) but usually do not seem to be significantly more efficacious than the original dopamine D2 receptor antagonists10. Although both conventional and atypical antipsychotics reliably reduce delusions and hallucinations, they have not enhanced functional recovery (for example, employment) for people with schizophrenia. One explanation is that the disability of schizophrenia is largely due to cognitive deficits, such as problems with attention and working memory, which these drugs fail to improve.
A focus on cognitive symptoms has led to a more recent hypothesis of schizophrenia as a ‘glutamate disorder’ (reviewed in ref. 11) Healthy volunteers given low doses of NMDA receptor antagonists, such as ketamine, manifest select aspects of schizophrenia, including some of the attentional and memory problems. Conversely, agents that modulate the glycine modulatory site on the NMDA receptor have been reported to reduce some of the cognitive symptoms of schizophrenia. The theory is that schizophrenia, particularly the cognitive symptoms of the disorder, may result from low activity of the NMDA receptor on GABA inhibitory interneurons in the prefrontal cortex.
Although there can be little argument that medications have transformed the treatment of psychosis, research focusing on the drugs instead of the illness has thus far yielded too little progress on the pathophysiology of schizophrenia. It is not clear, for instance, that either dopamine D2 receptors or interneuron NMDA receptors are related to the cause of this disorder. Although post-mortem studies have consistently reported a loss of GABA and reductions in key enzymes for glutamate biosynthesis, potentially consistent with the glutamate hypothesis, these changes may represent the effects of chronic illness or treatment of the disorder rather than the cause of schizophrenia11.
One approach that could separate cause from effect is genetics. Just as neuropharmacology dominated schizophrenia research in the late twentieth century, genetics has been a leading focus in the first decade of this century. Although in the ‘genomic era’ such a shift was inevitable, it was also presaged by a generation of twin and family studies demonstrating high heritability12,13. Reported concordance in monozygotic twins was roughly 50%, never the 100% figure one might expect for a Mendelian disorder, but considerably higher than dizygotic twins or siblings14.
High heritability has not, however, translated into a satisfying search for genetic lesions. Although early genome-wide or candidate-gene studies searching for common variants associated with schizophrenia were mostly disappointing, either because early findings failed to replicate or large-scale studies failed to detect genome-wide significance, recent international consortia combining single nucleotide polymorphism (SNP) data from several independent studies have found replicable associations with genes of the major histocompatability complex (MHC) region on chromosome 6p21.3-22.1, ZNF804A on chromosomes 2q32.1, neuregulin 1 (NRG1) on chromosome 8, as well as transcription factor 4 (TCF4) on 18q21.2 (refs 15–17). Other studies have reported SNPs in candidate genes associated either with schizophrenia or a broad phenotype of psychosis, notably for genes within the neuregulin–ERBB4 signalling pathway18, synaptic protein genes (for example, NRX1 (also known as PNO1))19, a potassium channel (KCNH2)20 and many other brain-expressed proteins (for example, dysbindin)21. Currently, at least 43 candidate genes have been identified, but individual effect sizes are consistently modest (http://www.schizophreniaforum.org/res/sczgene/TopResults.asp), especially relative to the evidence for high heritability22,23. Epistatic or additive effects of these variants may explain more of the risk, but results thus far on individual variants from case–control studies have not been useful for understanding an individual’s risk for schizophrenia.
In addition to the many reports of common single nucleotide variations, many rare structural genomic variants, such as copy number variants and translocations, have been described in schizophrenia (reviewed in ref. 24). These rare variants seem to have larger causative effects than previously reported SNPs, but most are not specific to schizophrenia and some occur only in a single family. The diversity and private nature of these mutations preclude a simple genetic explanation for schizophrenia, but these findings may yield important clues to pathophysiology. For instance, although the DISC1 translocation that confers very high risk for psychiatric disorder has been detected in only a single Scottish family, this private mutation has revealed important mechanisms of disease and identified a site where common variation may also confer risk (reviewed in ref. 25). Even more encouraging, the consistent reports that so many of these structural variants affect genes implicated in brain development may predict the future of schizophrenia research.
Mapping the pathophysiology of schizophrenia
A starting point for mapping the pathophysiology of schizophrenia can begin with the increasing recognition that this is a neurodevelopmental disorder, or perhaps more accurately a collection of neurodevelopmental disorders that involve alterations in brain circuits. Although Feinberg26, Weinberger27 and Murray28 proposed this approach more than two decades ago, the field is only now providing the evidence and recognizing the implications of shifting to a neurodevelopmental approach29,30.
Psychosis nearly always emerges in late adolescence or early adulthood, with a peak between ages 18 and 25, when the prefrontal cortex is still developing. We still do not understand all of the changes in normal or abnormal cortical development during this period. Attempts to map functional connectivity defined by imaging the default network demonstrate little integration until after age nine31. Longitudinal neuroimaging studies demonstrate changes in grey matter density until the mid-twenties with the prefrontal cortex being the last to mature32. The cellular basis for the observed reduction in grey-matter density with magnetic resonance imaging (MRI) is not clear although classical anatomical post-mortem studies indicate that both synaptic elimination and increased myelination continue into early adulthood33,34. Whereas the literature from human post-mortem neuroanatomy of adolescence is scant, studies in non-human primate brain demonstrate that the refinement of circuits during early adulthood includes pruning of asymmetric (excitatory) synapses, proliferation of inhibitory circuits and the continued elaboration of pyramidal dendrites as targets of inhibitory input35,36,37. Together these observations indicate that this late stage of brain maturation involves a careful calibration of excitatory–inhibitory balance in the cortex with the prefrontal cortex the last region to mature (Fig. 1). As one potentially relevant modulator of this balance, dopamine innervation of the prefrontal cortex increases markedly during adolescence38,39.
Figure 1: Neurodevelopmental model of schizophrenia.
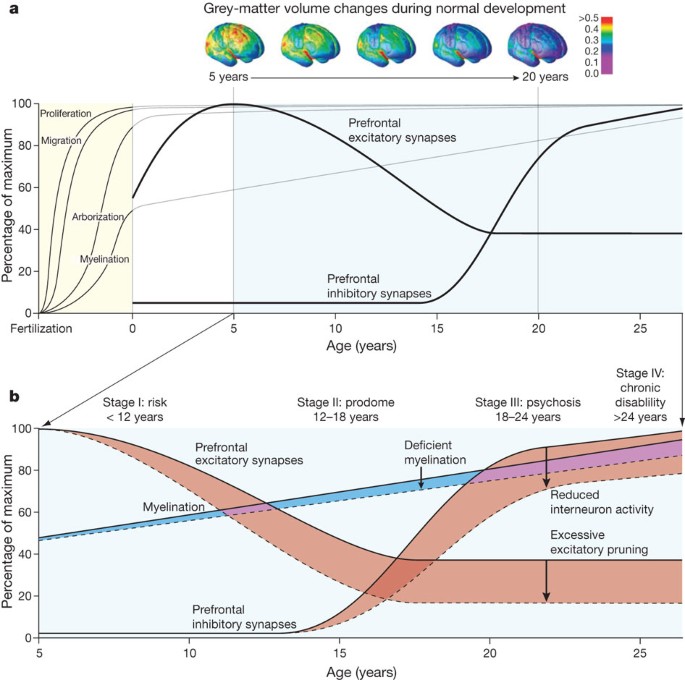
a, Normal cortical development involves proliferation, migration, arborization (circuit formation) and myelination, with the first two processes occurring mostly during prenatal life and the latter two continuing through the first two post-natal decades. The combined effects of pruning of the neuronal arbor and myelin deposition are thought to account for the progressive reduction of grey-matter volume observed with longitudinal neuroimaging. Beneath this observed overall reduction, local changes are far more complex. Data from human and non-human primate brain indicate increases in inhibitory and decreases in excitatory synaptic strength occurring in prefrontal cortex throughout adolescence and early adulthood, during the period of prodrome and emergence of psychosis. b, The trajectory in children developing schizophrenia could include reduced elaboration of inhibitory pathways and excessive pruning of excitatory pathways leading to altered excitatory–inhibitory balance in the prefrontal cortex. Reduced myelination would alter connectivity. Although some data support each of these possible neurodevelopmental mechanisms for schizophrenia, none has been proven to cause the syndrome. Detection of prodromal neurodevelopmental changes could permit early intervention with potential prevention or preemption of psychosis.
Although schizophrenic psychosis usually emerges between ages 18–25, several longitudinal population-based studies indicate that problems are evident much earlier. For instance, a recent report from a 45-year follow up of a Copenhagen birth cohort demonstrated that adults with schizophrenia have a history of delayed maturation including delayed developmental milestones in the first year40. Data from the Dunedin birth cohort, consistent with many previous studies41, indicated that IQ is reduced early and persistently in children destined to develop schizophrenia42. These precursors of schizophrenia are subtle and non-specific, but the consistency of the finding supports the hypothesis that psychosis does not emerge from a completely healthy brain.
The emerging picture from genetic studies also indicates that early brain development is affected. As noted earlier, many of the structural variants associated with schizophrenia implicate neurodevelopmental genes involved with neuronal proliferation, migration, or synapse formation43. Even genes that are not exclusively developmental seem to influence schizophrenia by their early disruption44. In a particularly intriguing example, Niwa et al.45 reported that a transient knockdown of DISC1 in the frontal cortex in the pre- and perinatal mouse brain led to neurochemical and behavioural disruptions emerging in early adulthood. Moreover, some of the vulnerability alleles of candidate genes, such as NRG1 and DISC1, seem to selectively influence splice variants expressed predominantly in developing cortex, implicating isoforms that show large developmental changes in expression in the prefrontal cortex46,47,48. As a final link to development, the genetics of schizophrenia overlaps with the genetics of autism and other neurodevelopmental disorders19,49. It is unclear why the same genetic variation associated with many different neurodevelopmental syndromes is manifested in some by age 3 years (autism) and in others after age 18 years (schizophrenia). Presumably there are genomic modifiers or possibly environmental influences that determine the specific syndrome. The study of discordant twins may yield important information for understanding the mismatches between genotype and phenotype.
Environmental factors identified so far have also implicated prenatal or perinatal events. Maternal malnutrition during famine50,51, infections in the second trimester52, perinatal injury53 and cytokine exposure54 have all been associated with subsequent increased risk for schizophrenia. Most of these effects are modest (less that twofold increase in risk) and none seem specific for schizophrenia, but in aggregate they demonstrate that early adverse experiences, including mid-gestational insults, are a risk factor for psychosis occurring two decades later. Gene-by-environment studies may demonstrate more robust effects55, but an even more promising approach may be epigenetic maps indicating the ‘scars’ of early experience or the stochastic changes emerging across development56. As an example, a gene disrupted by a rare copy number variant in autism was found to be repressed by hypermethylation in a large number of children with autism who had a perfectly normal genomic sequence57.
The model that emerges from this neurodevelopmental perspective is that of an early insult, a latent period through much of neural development, and the emergence of psychosis in late adolescence or early adulthood. One possibility is a lesion early in development that does not manifest until a much later developmental stage when compensatory changes can no longer suffice. Thompson and Levitt58 have called this developmental allostasis. A second, not mutually exclusive possibility is that the developmental lesion influences a pathway or a regulatory process, such as the fine tuning of excitatory and inhibitory synapses in the prefrontal cortex, which may have only subtle effects until a precise balance is required in late adolescence. Current data cannot distinguish between these two options, but either way a neurodevelopmental perspective implies the importance of timing and the opportunity for earlier intervention and prevention.
How will we map the trajectory of schizophrenia as a neurodevelopmental disease? Recent longitudinal studies of children with a rare, early-onset form of schizophrenia have used neuroimaging to identify differences in the trajectory of brain development. In these studies, children with schizophrenia seem to undergo excessive losses of grey matter and cortical thinning, essentially overshooting the normal pattern described earlier for adolescents59,60. These findings, although intriguing, are limited in that they do not reveal the changes before psychosis.
An opportunity for mapping earlier phases of the trajectory can be found in velocardiofacial syndrome, a syndrome associated with a microdeletion of chromosome 22q11 (reviewed in ref. 61). Approximately 30% of children with a microdeletion of 22q11 will develop a form of schizophrenia that clinically and neurocognitively cannot be distinguished from the idiopathic disorder62,63. Most of these children are detected as toddlers because of their cardiac disease. Important insights into the trajectory from risk to disorder may be gained from ongoing longitudinal studies of these children comparing cognitive, affective and neural development in those who do and do not develop psychosis among this cohort with a similar genomic deletion.
Will animal studies reveal the neurodevelopmental trajectory of schizophrenia? Unlike the many disorders in medicine that can be modelled in mice or flies, an animal model of schizophrenia seems unlikely. Indeed, aspects of the prefrontal neuroanatomy and the executive function deficits of schizophrenia seem to be distinctively human. This is not to say that studies in animals, especially non-human primates, will be unimportant for schizophrenia. We lack fundamental information on the normal development of the forebrain, from the timing and geography of gene expression to the patterns of circuit formation under various environmental conditions. With current technology, these critical developmental maps will only be derived from studies in animals. Animal studies can also aid the study of abnormal development. Whereas animal models of schizophrenia are not likely, ‘model animals’ such as mice and flies engineered with schizophrenia candidate genes will be highly informative for linking genetic variation to changes in cell and circuit function. For instance, mice with homologous deletions to the 22q11 lesion of velocardiofacial syndrome manifest differences in circuit formation and synaptic plasticity64,65. Such model animals will not only yield studies of disease mechanisms but opportunities for new treatment development.
Increasingly, however, it seems that humans may prove the best animal for modelling schizophrenia. Just as genes can create relevant models in non-human animals, genes can serve as a portal to mapping the pathophysiology of schizophrenia in cells from patients with the disorder. With induced pluripotent stem cells derived from fibroblasts of patients with schizophrenia, we should soon be able to study many different neural cell types, including their development, functional connections and response to perturbations. These cells do not need to recreate the disorder in a dish; they need only yield disordered molecular networks to reveal targets for developing new therapies. Through identifying new targets and high-throughput screening of existing small molecule libraries, we can expect the next generation of treatments for schizophrenia to be based on molecular pathophysiology rather than serendipity.
The stages of schizophrenia
Perhaps the most fundamental change from re-conceptualizing schizophrenia as a neurodevelopmental disorder is the notion of trajectory of illness. If the disorder begins in prenatal or perinatal life, then the psychosis of late adolescence must be seen not as the onset but as a late stage of the disorder. Indeed, we can begin to hypothesize four stages of schizophrenia, from risk to prodrome to psychosis to chronic disability66 (Table 1). At present, the diagnosis is based on the symptoms and signs of psychosis. With the advent of biomarkers and new cognitive tools as well as the identification of subtle clinical features, we are beginning to detect earlier stages of risk and prodrome.67
Table 1 Stages of schizophrenia
The earliest stage is risk, before detectable deficits. In 2010 we do not have the risk architecture of this syndrome, but we can begin to see some of the outlines, based on genomics. Beyond the rare, highly penetrant mutations (for example, DISC1 and the 22q11 deletion), epistatic interactions between more common, less penetrant variations may yield higher predictions of risk than our current list. Of course, the 50% concordance rate of homozygous twins reminds us that genomics will not predict all forms of risk. Identifying environmental factors, detecting critical epigenomic modifications, or mapping neural circuit differences may render more of the blueprint for risk, much as the algorithms for coronary artery disease use family history, plasma lipids and dietary history. The extent to which the risk factors for schizophrenia will be modifiable in the sense that we can reduce the risk for coronary artery disease or lung cancer remains to be seen. And although this earliest stage may not involve distress or help-seeking, longitudinal studies have begun to identify subtle but reproducible evidence for behavioural and cognitive problems in early childhood68,69,70. To define this earliest stage we will need to define the full architecture of individual risk: genetic and epigenetic biomarkers, cognitive indicators and physiological predictors of vulnerability to the disorder.
Over the past two decades, the pioneering work of McGorry and his colleagues71,72 has established the prodrome of schizophrenia as a valid second stage of the illness before psychosis. Whether defined as ultra-high risk or pre-psychosis, the prodrome is now identified based on changes in thoughts (for example, bizarre ideas falling short of psychotic ideation), social isolation and impaired functioning (for example, reduced school performance). Recognizing that these features might seem endemic to adolescence, the Structured Interview for Prodromal Syndromes (SIPS) was developed to distinguish high risk for psychosis from more common adolescent angst73. Recently a large multi-site project in the United States of 291 adolescents followed for 2.5 years reported that the prodrome represented a 405-fold increase in risk (relative to the general population) and that a combination of three factors (for example, genetic risk with recent functional decline, unusual thought content, and either suspicion/paranoia or reduced social functioning) resulted in a positive predictive power for conversion to psychosis of 74–81% (ref. 74). The addition of biomarkers, detected with functional or structural neuroimaging (reviewed in ref. 75), or the use of neuropsychological tests of reaction time or verbal memory76,77 may enhance detection and increase the predictive power. Given the high rate of behavioural distress in adolescence and the likelihood that many with prodromal symptoms will either mature out of them or develop other disorders, the challenge is to increase sensitivity for detecting ultra-high risk while not sacrificing specificity78. Specificity is a challenge: many of those who seek help for prodromal symptoms will develop other forms of psychopathology, not schizophrenia. What will we need to define this stage of schizophrenia? Although standardized clinical assessments will help and longitudinal imaging may yield biomarkers, it is likely that cognitive changes, such as reductions in working memory, may be the best predictor of the psychotic phase of schizophrenia79. Over the next few years, cognitive neuroscience will have a critical role in providing the tools for increasing the sensitivity and specificity of the schizophrenic prodrome80.
It is unclear to what extent intervening during the prodrome will either prevent or forestall psychosis. Results from single-site trials of atypical antipsychotics81, antidepressants82, lithium83 and cognitive behaviour therapy84 have had, at best, modest effects in reducing symptoms or preventing conversion to psychosis. A recent randomized double-blind placebo-controlled 12-week trial of long-chain omega-3 polyunsaturated fatty acids reported a 12-month conversion to psychosis in 2 of 41 (4.9%) individuals in the treated group versus 11 of 40 (27.5%) individuals in the placebo group85. Although promising, the overall rate of conversion (13 of 81) is lower than that observed in most prodromal cohorts. Current efforts to use cognitive remediation may identify a low-risk approach that could be used even if specificity were low86. An innovative, broad effort on youth mental health in Australia is addressing the issues of false positives, low specificity and potential stigma from early diagnosis by developing community-based, resilience-based interventions66.
Stage III of schizophrenia is psychosis manifested by hallucinations, delusions, disorganization of thought and behaviour, and psychomotor abnormalities. It is now clear that negative symptoms (loss of will, anhedonia, poverty of thought) and cognitive deficits (reduced working memory, poor cognitive control) are core features of the disorder that account for much of the long-term morbidity and poor functional outcomes87. Although the avolitional component of the disorder may define a special subgroup88, there is a new consensus that the negative symptoms and cognitive aspects of pathology are major unmet therapeutic needs89,90. If risk is analogous to hyperlipidemia, prodrome comparable to angina, then psychosis can be thought of as myocardial infarction with frequent residual loss of function. In spite of consistently positive acute responses to antipsychotic medications and psychosocial treatments, relapse rates approach 80% (ref. 2). Cognitive deficits and negative symptoms, whether preceding or emerging with psychosis seem, at best, only modestly responsive to current antipsychotic treatments91,92. The most urgent research priority in the near term will be effective treatments for the cognitive deficits, including the lack of insight that often inhibits adherence to both medication and psychosocial treatments.
Stage IV of schizophrenia involves chronic disability. In 1988, in the height of the AIDS epidemic, the editor of Nature noted that “schizophrenia is arguably the worst disease affecting mankind, even AIDS not excepted.”93 Not all individuals progress to this late stage of the illness, but for those who do the disability is not only psychiatric but medical. The oft-cited psychiatric deficits lead to unemployment, homelessness and incarceration, as noted earlier. A Finnish birth cohort study recently reported a 7% rate of suicide in schizophrenia, accounting for 50% of all deaths by age 39 (ref. 94). The medical complications of chronic schizophrenia are less well known. In 2010, smoking and obesity are epidemic among people with schizophrenia, with estimates of nicotine dependence ranging from 58–90% (ref. 95) and metabolic syndrome (obesity, hyperlipidemia, hyperglycemia and hypertension) present in 40% (ref. 96). Life expectancy for those with serious mental illness has been estimated at 56 years, approximately 25 years of premature mortality resulting usually from cardiopulmonary disease or other chronic medical conditions97. Importantly, many of the medical complications of schizophrenia can be prevented through tobacco cessation, dietary management and programs to manage cardiovascular health.
Schizophrenia in 2030
What is the prognosis of schizophrenia for 2030? I will venture a few predictions based on hope more than knowledge and recognizing that progress in understanding and treating schizophrenia may come from distant fields of science that have not yet been engaged in this area (Fig. 2).
Figure 2: A vision for schizophrenia over the next two decades.
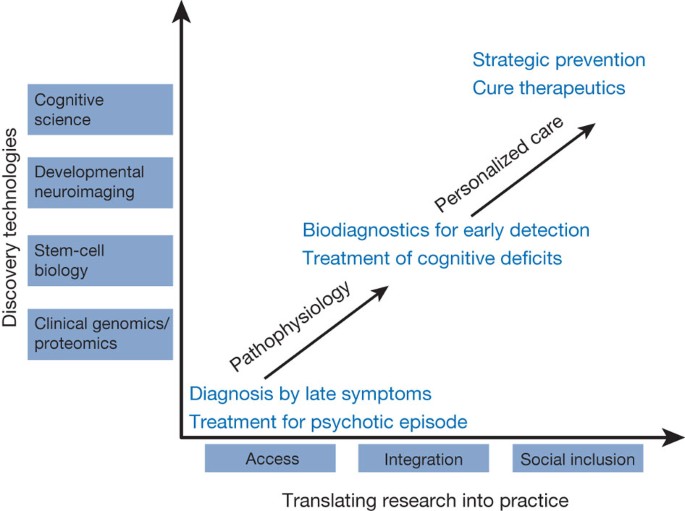
Currently diagnosis follows psychosis (stage III) and treatment focuses on reducing psychotic symptoms. The use of discovery technologies, which have already transformed the understanding and treatment of many other medical disorders, can transform our understanding of schizophrenia, yielding earlier diagnosis (stages I or II) with treatments focused on the cognitive deficits of this disorder. The ultimate goal, however, is cure and prevention based on an understanding of individual risk and the development of personalized care. In practice this means not only identifying risk and preemptive interventions but ensuring access to these interventions, integrating care and ensuring full social inclusion for people at any stage of the schizophrenia trajectory.
Prevention
Judging from the success of preventive approaches to cardiac death and disability, refocusing our approach to schizophrenia on early detection and early intervention could yield substantial improvements in outcomes over the next decade or two. This will, of course, require sensitive and specific diagnostic tools as well as safe and effective interventions. The diagnostic tools for schizophrenia, like the diagnostic tools for cardiovascular risk, will probably require a combination of approaches, including measures of genetic risk, imaging the efficiency of neural circuits, and, probably most specifically, early cognitive changes. Interventions that include an aggressive focus on cognition along with family support may prove surprisingly effective for preempting or forestalling psychosis. Although a ‘statin-like’ medication would be an unambiguous breakthrough, we should not assume that a medication will be more effective than harnessing the developing brain’s intrinsic plasticity for reversing the neural trajectory that leads from risk to prodrome. If the preemptive interventions are as effective as what we have today for coronary artery disease and if these are widely deployed, by 2030 we should expect a profound reduction in first-episode psychosis.
Reducing the cognitive deficits
The disability of schizophrenia in 2010 results more from the under-recognized and treatment-refractory cognitive deficits than from the more obvious and frequently treatable positive symptoms98. Over the next decade, potentially leveraging current research on cognition in Alzheimer’s disease, we can expect a series of pharmacological and nonpharmacological interventions that will reverse or mitigate the cognitive deficits of the disorder. Early initiation of these interventions will be transformative, but even in patients following psychosis, cognitive remediation may enhance employment, social inclusion and function in the community99. With interventions that reduce cognitive deficits, by 2030 we will be combining somatic, psychosocial and cognitive treatments with a goal of curing this disease for many patients.
Integration of care
One of the most egregious aspects of schizophrenia treatment in 2010 is the fragmentation of care, with medical care separated from psychiatric care and both isolated from psychosocial interventions, such as supportive employment and family education, which have a strong evidence base for effectiveness. Arguably, doing better with current treatments is our best short-term strategy for enhancing outcomes. A large multi-site effort in the United States, the Recovery After Initial Episode of Schizophrenia (RAISE) project, is developing a best-practices approach to bundled services that should provide some data about how much this can enhance outcomes. One can hope that in the near future, well before 2030, we will see all aspects of care being integrated in a continuous way, as is done increasingly for diabetes and other chronic disorders. Note, however, that the treatment of schizophrenia involves challenges not observed in most other chronic diseases. Denial of illness, paranoia, irrational thoughts, deficits in executive function and disruptive behaviour can all be part of the syndrome of untreated schizophrenia, complicating care for those with this disorder. Better treatments, not only better systems, will be necessary for better outcomes.
Stigma
Just as warehousing in institutions is mostly a memory today, imagine if the stigma associated with schizophrenia today were gone in 2030. In contrast to many other medical disorders, schizophrenia today too often defines a person rather than describing the illness. Our fear of psychosis or disruptive behaviour may keep us from seeing the heroic struggle that people with this disorder face just to survive amidst the internal chaos and panic that is part of this chronic illness. Our expectations of these citizens are low: they should stay out of jail, on their medications and not distress their families, friends and fellow citizens. They deserve better. As a vision for 2030, people who suffer from any stage of schizophrenia will be considered to be educable, employable and capable of living in intimate relationships with others.
Will we still use the term schizophrenia in 2030? The accumulating genomic evidence indicates that there may be scores or hundreds of lesions contributing to this final common syndrome. The clinical evidence supports the possibility that what we have labelled schizophrenia for the past century may be many different disorders with different outcomes88. And the stigma associated with the diagnosis, and the past history of misunderstanding and mistreatment also indicate that a change in the term may be advisable. In 2002, the Japanese terms for schizophrenia ‘Seishin-Bunretsu-Byo’ (‘mind-split disease’) was replaced officially by ‘Togo-Shitcho-Sho’ (‘integration disorder’)100. Some evidence indicates that this name change led to reduced stigma, in that fewer people associated the new name with criminality100.
Although semantic changes can be helpful, the transformations needed for those with this serious illness are likely to require not only a better label but better science (Fig. 2). In the next decade the challenge will be to integrate the impact of genetics, experience and development to identify a complete blueprint of the risk architecture of this syndrome. This should lead to a new taxonomy, identifying the many disorders within the syndrome we now call ‘schizophrenia’ and hopefully replacing this aggregate label with a series of more precise diagnoses based on pathophysiology. We need a personalized and preemptive approach, based on understanding and detecting individual risk and facilitated by safe and effective interventions for those in stages I and II of this disorder. In the meantime, we can create policies for social inclusion, family support and continuity of care to ensure that those in later stages of the syndrome have the best chance for recovery. Importantly, if recovery defined as a life in the community is our primary goal today, for 2030 our goals must include prevention, preemption and cure.
References
- Hegarty, J. D., Baldessarini, R. J., Tohen, M., Waternaux, C. & Oepen, G. One hundred years of schizophrenia: a meta-analysis of the outcome literature. Am. J. Psychiatry 151, 1409–1416 (1994)
CAS PubMed Google Scholar - Robinson, D. G., Woerner, M. G., McMeniman, M., Mendelowitz, A. & Bilder, R. M. Symptomatic and functional recovery from a first episode of schizophrenia or schizoaffective disorder. Am. J. Psychiatry 161, 473–479 (2004)
PubMed Google Scholar - Harrison, G. et al. Recovery from psychotic illness: a 15- and 25-year international follow-up study. Br. J. Psychiatry 178, 506–517 (2001)
CAS PubMed Google Scholar - Marwaha, S. et al. Rates and correlates of employment in people with schizophrenia in the UK, France and Germany. Br. J. Psychiatry 191, 30–37 (2007)
PubMed Google Scholar - Folsom, D. P. et al. Prevalence and risk factors for homelessness and utilization of mental health services among 10,340 patients with serious mental illness in a large public mental health system. Am. J. Psychiatry 162, 370–376 (2005)
PubMed Google Scholar - Kraepelin, E. Dementia Praecox and Paraphrenia (Chicago Medical Book Co., 1919)
Google Scholar - Bleuler, E. Dementia Praecox or the Group of Schizophrenias (International Universities Press, 1950)
Google Scholar - Arieti, S. Interpretation of Schizophrenia (Basic Books, 1974)
Google Scholar - Carlsson, A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1, 179–186 (1988)
CAS PubMed Google Scholar - Lieberman, J. A. et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 353, 1209–1223 (2005)Clinical results of the largest publically funded treatment trial for chronic schizophrenia demonstrates no significant advantage for second-generation anti-psychotic medications, except for clozaril.
CAS PubMed Google Scholar - Coyle, J. T. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 363–382 (2006)
Google Scholar - Kety, S. S. The significance of genetic factors in the etiology of schizophrenia: results from the national study of adoptees in Denmark. J. Psychiatr. Res. 21, 423–429 (1987)
CAS PubMed Google Scholar - McGuffin, P. & Gottesman, I. I. Risk factors for schizophrenia. N. Engl. J. Med. 341, 370–372 (1999)
CAS PubMed Google Scholar - Cardno, A. G. et al. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch. Gen. Psychiatry 56, 162–168 (1999)
CAS PubMed Google Scholar - Stefansson, H. et al. Common variants conferring risk of schizophrenia. Nature 460, 744–747 (2009)Largest meta-analysis of genomic variants in schizophrenia demonstrates genome-wide significance for three common variants as well as several rare copy number variants.
ADS CAS PubMed PubMed Central Google Scholar - The International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009)These results demonstrate similar genetic risk architecture for schizophrenia and bipolar disorder, indicating that the genetics of psychosis may not be specific to any of the currently identified psychiatric syndromes.
Google Scholar - Ettinger, U. et al. Prefrontal and striatal volumes in monozygotic twins concordant and discordant for schizophrenia. Schizophr. Bull. 10.1093/schbul/sbq060 (10 June 2010)
- Hahn, C. G. et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nature Med. 12, 824–828 (2006)
CAS PubMed Google Scholar - Rujescu, D. et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 18, 988–996 (2009)
CAS PubMed Google Scholar - Huffaker, S. J. et al. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nature Med. 15, 509–518 (2009)
CAS PubMed Google Scholar - Straub, R. E. et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 71, 337–348 (2002)
CAS PubMed PubMed Central Google Scholar - McClellan, J. & King, M. C. Genetic heterogeneity in human disease. Cell 141, 210–217 (2010)
CAS PubMed Google Scholar - Need, A. C. et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 5, e1000373 (2009)
PubMed PubMed Central Google Scholar - Bassett, A. S., Scherer, S. W. & Brzustowicz, L. M. Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. Am. J. Psychiatry 167, 899–914 (2010)
PubMed PubMed Central Google Scholar - Brandon, N. J. et al. Understanding the role of DISC1 in psychiatric disease and during normal development. J. Neurosci. 29, 12768–12775 (2009)
CAS PubMed PubMed Central Google Scholar - Feinberg, I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17, 319–334 (1982)
PubMed Google Scholar - Weinberger, D. R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 44, 660–669 (1987)
CAS PubMed Google Scholar - Murray, R. M., Jones, P. & O’Callaghan, E. Fetal brain development and later schizophrenia. Ciba Found. Symp. 156, 155–170 (1991)
CAS PubMed Google Scholar - Lewis, D. A. & Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 25, 409–432 (2002)
CAS PubMed Google Scholar - Jaaro-Peled, H. et al. Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1–ErbB4 and DISC1. Trends Neurosci. 32, 485–495 (2009)
CAS PubMed PubMed Central Google Scholar - Fair, D. A. et al. The maturing architecture of the brain’s default network. Proc. Natl Acad. Sci. USA 105, 4028–4032 (2008)
ADS CAS PubMed PubMed Central Google Scholar - Paus, T., Keshavan, M. & Giedd, J. N. Why do many psychiatric disorders emerge during adolescence? Nature Rev. Neurosci. 9, 947–957 (2008)
CAS Google Scholar - Huttenlocher, P. R. Synapse elimination and plasticity in developing human cerebral cortex. Am. J. Ment. Defic. 88, 488–496 (1984)
CAS PubMed Google Scholar - Yakovlev, P. & Lecours, A. R. in Regional Development of the Brain in Early Life (ed. Minkowski, A.) (Blackwell Scientific, 1967)
Google Scholar - Rakic, P., Bourgeois, J. P., Eckenhoff, M. F., Zecevic, N. & Goldman-Rakic, P. S. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232, 232–235 (1986)
ADS CAS PubMed Google Scholar - Hashimoto, T. et al. Protracted developmental trajectories of GABAA receptor α1 and α2 subunit expression in primate prefrontal cortex. Biol. Psychiatry 65, 1015–1023 (2009)
CAS PubMed PubMed Central Google Scholar - Lewis, D. A. & Gonzalez-Burgos, G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology 33, 141–165 (2008)
PubMed Google Scholar - Rosenberg, D. R. & Lewis, D. A. Postnatal maturation of the dopaminergic innervation of monkey prefrontal and motor cortices: a tyrosine hydroxylase immunohistochemical analysis. J. Comp. Neurol. 358, 383–400 (1995)
CAS PubMed Google Scholar - Lambe, E. K., Krimer, L. S. & Goldman-Rakic, P. S. Differential postnatal development of catecholamine and serotonin inputs to identified neurons in prefrontal cortex of rhesus monkey. J. Neurosci. 20, 8780–8787 (2000)
CAS PubMed PubMed Central Google Scholar - Sørensen, H. J. et al. Early developmental milestones and risk of schizophrenia: a 45-year follow-up of the Copenhagen Perinatal Cohort. Schizophr. Res. 118, 41–47 (2010)
PubMed PubMed Central Google Scholar - Woodberry, K. A., Giuliano, A. J. & Seidman, L. J. Premorbid IQ in schizophrenia: a meta-analytic review. Am. J. Psychiatry 165, 579–587 (2008)
PubMed Google Scholar - Reichenberg, A. et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am. J. Psychiatry 167, 160–169 (2010)
PubMed PubMed Central Google Scholar - Walsh, T. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543 (2008)
ADS CAS PubMed Google Scholar - Li, W. et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc. Natl Acad. Sci. USA 104, 18280–18285 (2007)
ADS CAS PubMed PubMed Central Google Scholar - Niwa, M. et al. Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65, 480–489 (2010)Mouse study uses transient knockdown of DISC1 in forebrain to show the unique developmental effect of this protein, with biochemical, physiological and behavioural changes emerging after puberty.
CAS PubMed PubMed Central Google Scholar - Tan, W. et al. Molecular cloning of a brain-specific, developmentally regulated neuregulin 1 (NRG1) isoform and identification of a functional promoter variant associated with schizophrenia. J. Biol. Chem. 282, 24343–24351 (2007)
CAS PubMed Google Scholar - Colantuoni, C. et al. Age-related changes in the expression of schizophrenia susceptibility genes in the human prefrontal cortex. Brain Struct. Funct. 213, 255–271 (2008)
PubMed Google Scholar - Nakata, K. et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proc. Natl Acad. Sci. USA 106, 15873–15878 (2009)Study of DISC1 shows isoforms expressed predominantly during human brain development are influenced by alleles associated with schizophrenia.
ADS CAS PubMed PubMed Central Google Scholar - Guilmatre, A. et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 66, 947–956 (2009)
CAS PubMed PubMed Central Google Scholar - Susser, E. S. & Lin, S. P. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944–1945. Arch. Gen. Psychiatry 49, 983–988 (1992)
CAS PubMed Google Scholar - St Clair, D. et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. J. Am. Med. Assoc. 294, 557–562 (2005)
CAS Google Scholar - Brown, A. S. & Derkits, E. J. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280 (2010)
PubMed PubMed Central Google Scholar - Cannon, M., Jones, P. B. & Murray, R. M. Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092 (2002)
PubMed Google Scholar - Ellman, L. M. et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin-8. Schizophr. Res. 121, 46–54 (2010)
PubMed PubMed Central Google Scholar - Nicodemus, K. K. et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol. Psychiatry 13, 873–877 (2008)
CAS PubMed Google Scholar - Feinberg, A. P. & Irizarry, R. A. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc. Natl Acad. Sci. USA 107 (suppl. 1). 1757–1764 (2010)
ADS CAS PubMed Google Scholar - Gregory, S. G. et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7, 62 (2009)Novel observation of hypermethylation of candidate gene in autism indicates potential mechanism by which rare structural variants can reveal sites for epigenetic modification leading to changes in gene expression.
PubMed PubMed Central Google Scholar - Thompson, B. L. & Levitt, P. Now you see it, now you don’t—closing in on allostasis and developmental basis of psychiatric disorders. Neuron 65, 437–439 (2010)
CAS PubMed Google Scholar - Gogtay, N. et al. Comparison of progressive cortical gray matter loss in childhood-onset schizophrenia with that in childhood-onset atypical psychoses. Arch. Gen. Psychiatry 61, 17–22 (2004)First demonstration of the specificity of cortical changes in childhood-onset schizophrenia, providing evidence for excessive loss of gray matter in prefrontal cortex.
PubMed Google Scholar - Rapoport, J. L. et al. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry 56, 649–654 (1999)
CAS PubMed Google Scholar - Karayiorgou, M., Simon, T. J. & Gogos, J. A. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nature Rev. Neurosci. 11, 402–416 (2010)Important, comprehensive review of basic and clinical research on 22q11 microdeletions, providing a pathway for linking genetic lesions to the pathophysiology of schizophrenia.
CAS Google Scholar - Bassett, A. S. et al. The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry 160, 1580–1586 (2003)
PubMed PubMed Central Google Scholar - Chow, E. W., Watson, M., Young, D. A. & Bassett, A. S. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr. Res. 87, 270–278 (2006)
PubMed PubMed Central Google Scholar - Mukai, J. et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature Neurosci. 11, 1302–1310 (2008)
CAS PubMed Google Scholar - Stark, K. L. et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nature Genet. 40, 751–760 (2008)
CAS PubMed Google Scholar - McGorry, P. D., Yung, A. R., Bechdolf, A. & Amminger, P. Back to the future: predicting and reshaping the course of psychotic disorder. Arch. Gen. Psychiatry 65, 25–27 (2008)
PubMed Google Scholar - Nestler, E. J. & Hyman, S. E. Animal models of neuropsychiatric disorders. 13, 1161–1169 (2010)
- Cannon, T. D. et al. Childhood cognitive functioning in schizophrenia patients and their unaffected siblings: a prospective cohort study. Schizophr. Bull. 26, 379–393 (2000)
CAS PubMed Google Scholar - Cornblatt, B., Obuchowski, M., Roberts, S., Pollack, S. & Erlenmeyer-Kimling, L. Cognitive and behavioral precursors of schizophrenia. Dev. Psychopathol. 11, 487–508 (1999)
CAS PubMed Google Scholar - Erlenmeyer-Kimling, L. et al. Attention, memory, and motor skills as childhood predictors of schizophrenia-related psychoses: the New York High-Risk Project. Am. J. Psychiatry 157, 1416–1422 (2000)Classic study identifying childhood predictors of adult psychosis in longitudinal design.
CAS PubMed Google Scholar - Henry, L. P. et al. The EPPIC follow-up study of first-episode psychosis: longer-term clinical and functional outcome 7 years after index admission. J. Clin. Psychiatry 71, 716–728 (2010)
PubMed Google Scholar - Yung, A. R. et al. Validation of “prodromal” criteria to detect individuals at ultra high risk of psychosis: 2 year follow-up. Schizophr. Res. 105, 10–17 (2008)
PubMed Google Scholar - Woods, S. W. et al. Validity of the prodromal risk syndrome for first psychosis: findings from the North American Prodrome Longitudinal Study. Schizophr. Bull. 35, 894–908 (2009)
PubMed PubMed Central Google Scholar - Cannon, T. D. et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch. Gen. Psychiatry 65, 28–37 (2008)Multi-site study to validate the prodrome of schizophrenia, identifying predictors of conversion to psychosis.
PubMed PubMed Central Google Scholar - Smieskova, R. et al. Neuroimaging predictors of transition to psychosis—a systematic review and meta-analysis. Neurosci. Biobehav. Rev. 34, 1207–1222 (2010)
CAS PubMed Google Scholar - Cohen, J. D., Barch, D. M., Carter, C. & Servan-Schreiber, D. Context-processing deficits in schizophrenia: converging evidence from three theoretically motivated cognitive tasks. J. Abnorm. Psychol. 108, 120–133 (1999)
CAS PubMed Google Scholar - Seidman, L. J. et al. Neuropsychology of the prodrome to psychosis in the NAPLS consortium: relationship to family history and conversion to psychosis. Arch. Gen. Psychiatry 67, 578–588 (2010)
PubMed PubMed Central Google Scholar - Riecher-Rössler, A. et al. Efficacy of using cognitive status in predicting psychosis: a 7-year follow-up. Biol. Psychiatry 66, 1023–1030 (2009)
PubMed Google Scholar - Kremen, W. S. et al. Cognitive decline in schizophrenia from childhood to midlife: a 33-year longitudinal birth cohort study. Schizophr. Res. 118, 1–5 (2010)
PubMed PubMed Central Google Scholar - Carter, C. S., Barch, D. M., Gur, R., Pinkham, A. & Ochsner, K. CNTRICS final task selection: social cognitive and affective neuroscience-based measures. Schizophr. Bull. 35, 153–162 (2009)
PubMed Google Scholar - Woods, S. W. et al. Randomized trial of olanzapine versus placebo in the symptomatic acute treatment of the schizophrenic prodrome. Biol. Psychiatry 54, 453–464 (2003)
CAS PubMed Google Scholar - Cornblatt, B. A. et al. Can antidepressants be used to treat the schizophrenia prodrome? Results of a prospective, naturalistic treatment study of adolescents. J. Clin. Psychiatry 68, 546–557 (2007)
CAS PubMed Google Scholar - Berger, G. et al. Neuroprotection in emerging psychotic disorders. Early Interv. Psychiatry 1, 114–127 (2007)
Google Scholar - Morrison, A. P. et al. Cognitive therapy for the prevention of psychosis in people at ultra-high risk: randomised controlled trial. Br. J. Psychiatry 185, 291–297 (2004)
PubMed Google Scholar - Amminger, G. P. et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch. Gen. Psychiatry 67, 146–154 (2010)
CAS PubMed Google Scholar - Keefe, R. S. et al. Report from the Working Group Conference on multisite trial design for cognitive remediation in schizophrenia. Schizophr. Bull. 10.1093/schbul/sbq010 (1 March 2010)
- Hyman, S. E. & Fenton, W. S. Medicine. What are the right targets for psychopharmacology? Science 299, 350–351 (2003)
CAS PubMed Google Scholar - Kirkpatrick, B., Buchanan, R. W., Ross, D. E. & Carpenter, W. T., Jr A separate disease within the syndrome of schizophrenia. Arch. Gen. Psychiatry 58, 165–171 (2001)
CAS PubMed Google Scholar - Buchanan, R. W. et al. A summary of the FDA-NIMH-MATRICS workshop on clinical trial design for neurocognitive drugs for schizophrenia. Schizophr. Bull. 31, 5–19 (2005)
PubMed Google Scholar - Kirkpatrick, B., Fenton, W. S., Carpenter, W. T., Jr & Marder, S. R. The NIMH-MATRICS consensus statement on negative symptoms. Schizophr. Bull. 32, 214–219 (2006)
PubMed PubMed Central Google Scholar - Keefe, R. S. et al. Effects of olanzapine, quetiapine, and risperidone on neurocognitive function in early psychosis: a randomized, double-blind 52-week comparison. Am. J. Psychiatry 164, 1061–1071 (2007)
PubMed Google Scholar - Swartz, M. S. et al. Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. Am. J. Psychiatry 164, 428–436 (2007)
PubMed Google Scholar - Editor Where next with psychiatric illness? Nature 336, 95–96 (1988)
Google Scholar - Alaräisänen, A. et al. Suicide rate in schizophrenia in the Northern Finland 1966 Birth Cohort. Soc. Psychiatry Psychiatr. Epidemiol. 44, 1107–1110 (2009)
PubMed Google Scholar - Kelly, D. L. et al. Cigarette smoking and mortality risk in people with schizophrenia. Schizophr. Bull. 10.1093/schbul/sbp152 (17 December 2009)
- McEvoy, J. P. et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr. Res. 80, 19–32 (2005)
PubMed Google Scholar - Colton, C. W. & Manderscheid, R. W. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Prev. Chronic Dis. 3, A42 (2006)
PubMed PubMed Central Google Scholar - Harvey, P. D., Reichenberg, A., Bowie, C. R., Patterson, T. L. & Heaton, R. K. The course of neuropsychological performance and functional capacity in older patients with schizophrenia: influences of previous history of long-term institutional stay. Biol. Psychiatry 67, 933–939 (2010)
PubMed PubMed Central Google Scholar - Bell, M. D., Zito, W., Greig, T. & Wexler, B. E. Neurocognitive enhancement therapy with vocational services: work outcomes at two-year follow-up. Schizophr. Res. 105, 18–29 (2008)
PubMed Google Scholar - Takahashi, H. et al. Impact of changing the Japanese term for “schizophrenia” for reasons of stereotypical beliefs of schizophrenia in Japanese youth. Schizophr. Res. 112, 149–152 (2009)
PubMed Google Scholar
Acknowledgements
The author appreciates comments on this manuscript from C. Carter, W. Carpenter, H. Heimer, D. Lewis and D. Weinberger.
Author information
Authors and Affiliations
- National Institute of Mental Health, Bethesda, 20892, Maryland, USA
Thomas R. Insel
Authors
- Thomas R. Insel
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toThomas R. Insel.
Ethics declarations
Competing interests
The author declares no competing financial interests.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Insel, T. Rethinking schizophrenia.Nature 468, 187–193 (2010). https://doi.org/10.1038/nature09552
- Published: 10 November 2010
- Issue Date: 11 November 2010
- DOI: https://doi.org/10.1038/nature09552
Editorial Summary
Schizophrenia today: three views of the future
Three Perspectives in this issue cover different aspects of the current state of our knowledge about schizophrenia. Thomas Insel, director of the US National Institute of Mental Health in Bethesda, Maryland, outlines a new approach to schizophrenia that could in time lead to new treatments. He calls for schizophrenia to be emphasized as a neurodevelopmental disorder in which psychosis is a late — and potentially curable — stage. Andreas Meyer-Lindenberg, director of the Central Institute of Mental Health in Mannheim, Germany, explains how neuroimaging and other systems-level techniques can help develop future treatment. And Jim van Os, Gunter Kenis and Bart Rutten review our knowledge of the environmental factors that influence schizophrenia risk, and the major challenges that will be involved in teasing them out.