Increased Diels-Alderase activity through backbone remodeling guided by Foldit players (original) (raw)
- Letter
- Published: 22 January 2012
- Justin B Siegel1 na1,
- Jacob B Bale1,2,
- Seth Cooper3,
- Firas Khatib1,
- Betty W Shen4,
- Foldit Players1,
- Barry L Stoddard4,
- Zoran Popovic3 &
- …
- David Baker1,5
Nature Biotechnology volume 30, pages 190–192 (2012) Cite this article
- 8645 Accesses
- 257 Citations
- 192 Altmetric
- Metrics details
Subjects
Abstract
Computational enzyme design holds promise for the production of renewable fuels, drugs and chemicals. De novo enzyme design has generated catalysts for several reactions, but with lower catalytic efficiencies than naturally occurring enzymes1,2,3,4. Here we report the use of game-driven crowdsourcing to enhance the activity of a computationally designed enzyme through the functional remodeling of its structure. Players of the online game Foldit5,6 were challenged to remodel the backbone of a computationally designed bimolecular Diels-Alderase3 to enable additional interactions with substrates. Several iterations of design and characterization generated a 24-residue helix-turn-helix motif, including a 13-residue insertion, that increased enzyme activity >18-fold. X-ray crystallography showed that the large insertion adopts a helix-turn-helix structure positioned as in the Foldit model. These results demonstrate that human creativity can extend beyond the macroscopic challenges encountered in everyday life to molecular-scale design problems.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout
Additional access options:
Figure 1: Crystal structure of Foldit design is similar to player model.
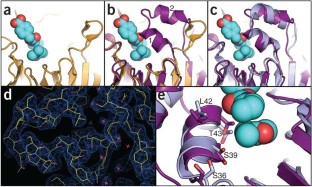
The alternative text for this image may have been generated using AI.
Figure 2: Activity of designed enzymes DA_20_10, CE0, CE4 and CE6.

The alternative text for this image may have been generated using AI.
Similar content being viewed by others
Accession codes
Accessions
Protein Data Bank
References
- Rothlisberger, D. et al. Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195 (2008).
Article Google Scholar - Jiang, L. et al. De novo computational design of retro-aldol enzymes. Science 319, 1387–1391 (2008).
Article CAS Google Scholar - Siegel, J.B. et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010).
Article CAS Google Scholar - Bar-Even, A. et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011).
Article CAS Google Scholar - Khatib, F. et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat. Struct. Mol. Biol. 18, 1175–1177 (2011).
Article CAS Google Scholar - Cooper, S. et al. Predicting protein structures with a multiplayer online game. Nature 466, 756–760 (2010).
Article CAS Google Scholar - Sterner, R. & Hocker, B. Catalytic versatility, stability, and evolution of the (βα)8-barrel enzyme fold. Chem. Rev. 105, 4038–4055 (2005).
Article CAS Google Scholar - Hu, X., Wang, H., Ke, H. & Kuhlman, B. High-resolution design of a protein loop. Proc. Natl. Acad. Sci. USA 104, 17668–17673 (2007).
Article CAS Google Scholar - Murphy, P.M., Bolduc, J.M., Gallaher, J.L., Stoddard, B.L. & Baker, D. Alteration of enzyme specificity by computational loop remodeling and design. Proc. Natl. Acad. Sci. USA 106, 9215–9220 (2009).
Article CAS Google Scholar - Havranek, J.J. & Baker, D. Motif-directed flexible backbone design of functional interactions. Protein Sci. 18, 1293–1305 (2009).
Article CAS Google Scholar - Park, H.S. et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science 311, 535–538 (2006).
Article CAS Google Scholar - Religa, T.L. et al. The helix-turn-helix motif as an ultrafast independently folding domain: the pathway of folding of Engrailed homeodomain. Proc. Natl. Acad. Sci. USA 104, 9272–9277 (2007).
Article CAS Google Scholar - Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Article Google Scholar - The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
- Kunkel, T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82, 488–492 (1985).
Article CAS Google Scholar - Siegel, J.B. et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010).
Article CAS Google Scholar - Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods In Enzymology: Macromolecular Crystallography. Part A 276, 307–326 (1997).
Article CAS Google Scholar - McCoy, A.J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Article CAS Google Scholar - Collaborative Computational Project, Number 4. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
- Scharff, E.I., Koepke, J., Fritzsch, G., Luecke, C. & Rueterjans, H. Crystal structure of diisopropylfluorophosphatase from Loligo vulgaris. Structure 9, 493–502 (2001).
Article CAS Google Scholar - Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Article Google Scholar - Krissinel, E. & Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 (2004).
Article CAS Google Scholar - Murshudov, G.N., Vagin, A.A. & Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997).
Article CAS Google Scholar - Winn, M.D., Isupov, M.N. & Murshudov, G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 (2001).
Article CAS Google Scholar - Vagin, A.A. et al. Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 (2004).
Article Google Scholar - Laskowski, R.A., Macarthur, M.W., Moss, D.S. & Thornton, J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993).
Article CAS Google Scholar
Acknowledgements
We would like to acknowledge the members of the Foldit team for their help designing and developing the game and all the Foldit players who volunteered to make this work possible. We would also like to thank J. Thompson for useful scripts, as well as B. Siegel and M. Eiben for helpful comments on the manuscript. This work was supported by the Center for Game Science at the University of Washington, US Defense Advanced Research Projects Agency (DARPA) grant N00173-08-1-G025, the DARPA PDP program, the Howard Hughes Medical Institute (D.B.), a National Science Foundation graduate research fellowship to J.B.B. (grant no. DGE-0718124), and a National Science Foundation grant for F.K. (grant no. 0906026).
Author information
Author notes
- Christopher B Eiben and Justin B Siegel: These authors contributed equally to this work.
Authors and Affiliations
- Department of Biochemistry, University of Washington, Seattle, Washington, USA
Christopher B Eiben, Justin B Siegel, Jacob B Bale, Firas Khatib, Foldit Players & David Baker - Graduate Program in Molecular and Cellular Biology, University of Washington, Seattle, Washington, USA
Jacob B Bale - Department of Computer Science and Engineering, University of Washington, Seattle, Washington, USA
Seth Cooper & Zoran Popovic - Division of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, Washington, USA
Betty W Shen & Barry L Stoddard - Howard Hughes Medical Institute, University of Washington, Seattle, Washington, USA
David Baker
Authors
- Christopher B Eiben
- Justin B Siegel
- Jacob B Bale
- Seth Cooper
- Firas Khatib
- Betty W Shen
- Foldit Players
- Barry L Stoddard
- Zoran Popovic
- David Baker
Contributions
C.B.E. analyzed community models, in addition to designing, building and testing the enzyme libraries. J.B.S. designed the experimental and computational methods, and built the initial computational models. F.K. set up the Foldit puzzles and curated the player results for analysis by C.B.E. S.C. led design and development of Foldit. B.L.S., J.B.B. and B.W.S. grew the crystals and collected X-ray diffraction data, processed X-ray data and analyzed the structure. Foldit Players designed new protein backbones and sequences. Z.P. and D.B. contributed to the writing of the manuscript.
Corresponding author
Correspondence toDavid Baker.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Eiben, C., Siegel, J., Bale, J. et al. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players.Nat Biotechnol 30, 190–192 (2012). https://doi.org/10.1038/nbt.2109
- Received: 25 October 2011
- Accepted: 04 January 2012
- Published: 22 January 2012
- Issue date: February 2012
- DOI: https://doi.org/10.1038/nbt.2109