Neoantigen prediction and the need for validation (original) (raw)
To the Editor:
The Editorial in your February issue entitled “The problem with neoantigen prediction” highlighted some of the challenges in identifying and validating cancer neoantigens—tumor-specific antigens—for personalized immunotherapy. This timely Editorial discussed some of the computational methods currently used to predict which somatic DNA mutations could give rise to neoantigens capable of eliciting an effective anti-tumor T-cell response when administered to patients. The article concluded that more research needs to be done before neoepitope prediction and validation becomes routine and personalized immunotherapy, a clinical reality.
Here, we wish to bring attention to the crucial need for experimental validation of mutant peptides (neopeptides) predicted to bind to major histocompatibility complex (MHC). Only a small subset of these peptides will be processed and presented in the context of MHC on the cell surface. And only a subset of those will be 'neoepitopes'—recognized by a T-cell receptor (TCR)-bearing T cell and, as a result, potentially immunogenic. Experimental validation of therapeutically relevant immunogenicity is a crucial step in improving the odds of successful immunotherapy.
Over the past several decades, thousands of patients have been vaccinated with tumor-associated antigens (antigens overexpressed by cancer cells or embryonic antigens reexpressed by cancer cells). Because such antigens were recognized by T cells from patients that cleared the tumor1, the hope has been that universal vaccines could be developed for specific cancers. For the most part, however, vaccination with such antigens has been ineffective. Recent work has shown that the T-cell repertoires of some cancer patients treated with immune checkpoint inhibitors, such as anti-CTLA-4 (cytotoxic T-lymphocyte antigen 4) and anti-PD1 (programmed death receptor 1)/PD-L1, contain neoepitope-specific T cells, refocusing attention on neoantigens as potential cancer vaccines2,3,4. Genomic sequencing and bioinformatics provide formidable tools for the identification of tumor-specific non-synonymous mutations, frameshift mutations, and gene rearrangements from which to select tumor proteins and peptides for immunotherapy.
As summarized in the Editorial, in silico methods aim to identify mutant peptides—likely processed by the tumor cell into short peptides—that bind the patient's MHC class I/II molecules, and that may contact a TCR and ultimately prove immunogenic. The identification of putative neoepitopes proceeds from DNA and/or RNA sequencing to predicting the MHC binding of mutation-containing 8- to 15-amino-acid peptide sequences. Processing can be predicted by the existing algorithms, but prediction is preferably done by direct identification of peptides bound to the MHC in the tumor by elution followed by mass spectrometry (MS)5. However, the MS approach is fraught with sensitivity issues and the likelihood of missing important epitopes; at present there is no single high-throughput method that allows for a comprehensive and certain identification of putative neoepitopes6. Recognition of naturally processed peptides by CD8 T-cell killing remains the most sensitive and accurate method because a single MHC–peptide complex suffices to mediate both recognition and killing by the T cell7. For instance, one could use T cells generated in HLA transgenic mice immunized with the reference neopeptide. Computational methods can be improved to more accurately predict MHC binding and processing and to predict in general terms antigenicity (e.g., the presence of aromatic amino acids and residues with bulky side chains8). These predictions lighten the burden of immunogenicity testing by reducing the number of candidate peptides, but establishing immunogenicity through empirical experimentation, although time- and resource-consuming, remains a necessary step in developing personalized immunotherapy.
Data obtained from large-scale analysis of peptides from complex viruses (dengue and vaccinia) predict that only ∼1% will bind MHC (depending on the accuracy of computational tools for a given MHC allele, the range is from 0.07% to 10%)9,10. Of the ∼1% binding MHC, only ∼50% will be recognized by a T cell, but only 30–40% are naturally processed, enabling target cell killing10 (Fig. 1). In a report that systematically interrogated patient responses to vaccination with predicted tumor neoepitopes, three melanoma patients were each immunized with seven peptides with _in vitro_-corroborated MHC-binding affinities <500 nM11. Of the 21 peptides tested, only 9 induced a T-cell response. Three of the nine neoepitopes were 'dominant' (the responding T cells were present in the patient before immunization), four were 'subdominant' (the T cells were induced by neopeptide immunization), and two were 'cryptic' (the responding T cells reacted to the neoepitopes but not to cells expressing the corresponding peptides). Thus, only 30% of the tested peptides (7 peptides) elicited a T-cell response in vivo, which is intriguingly similar to the findings with viral peptides.
Figure 1: Selection and validation of mutant peptides in genomic-based personalized immunotherapy.
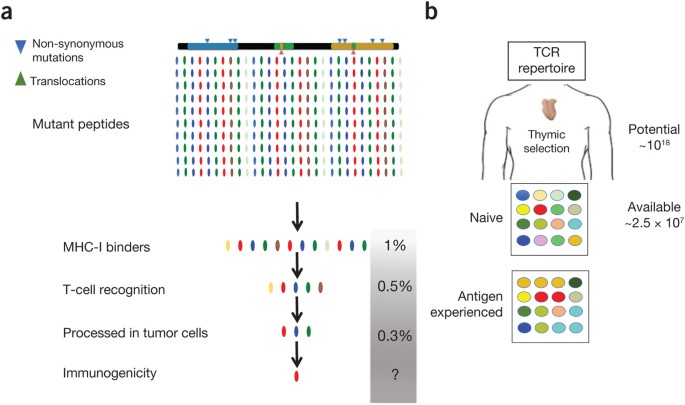
(a) Bioinformatics drives the selection of candidate neoepitopes from genomic sequencing data of the patient's tumor. In the case of MHC-I, a >99% reduction in the number of putative peptides occurs after filtering for MHC binding, potential TCR contacts, and processing in tumor cells. Peptides that survive the selection process need to be validated experimentally for immunogenicity. (b) The immunogenicity of putative neoepitopes is subject to limitations intrinsic to the available T-cell repertoire of the patient, whose main determinants are size and TCR diversity, which also reflects skewing by the antigenic experiences (e.g., infections) of the individual. MHC binder candidate neoepitopes: all mutant peptides predicted to bind any MHC-I allele with measurable affinity. These average 1% of all mutant peptides per allele, assuming six MHC-I alleles (HLA-A, B, and C) and no allelic competition.
Although binding to MHC-I is currently the most effective computational filter for removing nonantigenic peptides12, methods that identify competing MHC alleles can also reduce the burden of experimental validation. Humans express 12 MHC alleles: 6 class I (HLA-A, B, and C) and 6 class II (HLA-DR, DP, and DQ). However, as different alleles compete for peptides13,14 mono-allelic profiling of the immunopeptidome15 may not recapitulate the fate of a single peptide within the complexity of the cell haplotype in vivo and computational methods may need to be developed to allow for the complexity of the system.
We must also ask whether our knowledge of cancer biology could be exploited for in silico screening. The majority of tumors are highly heterogeneous, and distinct regions of a single lesion can have different mutational profiles. Should we limit selection to peptides resulting from clonal mutations (present in all cancer cells in the tumor) or include subclonal mutations (present in only a subset of cells)? Evidence from non-small-cell lung cancer and melanoma patients treated with immune checkpoint inhibitors suggested that only T-cell responses to clonal neoepitopes were associated with clinical benefit and prolonged survival16. Thus, cancer genomic computational methods could guide the work of immunologists accordingly by focusing on mutations that have a variant allele fraction of 50% (i.e., 100% of heterozygous mutations).
Another key question is whether the same algorithms should be applied for cancers where the frequency of non-synonymous mutations differs markedly17. We suggest that, for high-mutational-burden tumors (e.g., melanoma, lung adenocarcinoma, and bladder carcinoma), algorithms should be used to narrow down the number of candidate neoepitopes, followed by experimental validation with emphasis on peptides of clonal origin. For cancers with low mutational burden (e.g., glioblastoma and acute myeloid leukemia), all predicted neoepitopes should be experimentally validated. An additional consideration is whether neoepitopes that induce a cross-reactive T-cell response to the wild-type antigen should be considered for vaccine development16. Arguably, whereas this would be advantageous if the wild-type antigen is expressed only by cancer cells (e.g., cancer-specific telomerase reverse transcriptase and MAGE), it could cause adverse events if the antigen is expressed on normal somatic cells.
Clearly, the patient's existing T-cell repertoire determines the ability to generate an anti-tumor T-cell response. The complex process of selecting T cells restricted to self-MHC but tolerant to self-antigens occurs during ontogeny and is regulated by spatial, quantitative, and qualitative aspects of self-recognition in the thymic microenvironment18. In theory, ∼1018 TCR specificities could be generated by recombination of human TCR genes; however, the peripheral repertoire actually contains only ∼2.5 × 107 clonotypes19 (Fig. 1b). To this, we must factor in the individual's genetic and environmental experience, which further shapes the available T-cell repertoire. This influence is best exemplified by a large-scale study of monozygotic twins discordant for cytomegalovirus status, in which differences in immunological parameters were largely determined by non-heritable factors20. The presence of a tumor could certainly limit the available tumor-specific TCR repertoire. We therefore argue that, at present, the composition and specificity of the available TCR repertoire cannot be determined by an in silico approach.
Lastly, we note the relative dearth of in silico methods for predicting MHC-II-restricted putative neoepitopes. Activation and maintenance of a CD8+ T-cell response is dependent on concomitant activation of MHC-II-restricted helper T cells during priming21. Consistent with this, CD4+ T cells are known to play crucial roles in the anti-tumor response in vivo, and many reports have established that MHC-II-restricted neoepitopes can be immunogenic and elicit anti-tumor protection21. Indeed, peripheral CD4+ T cells are substantially expanded in some patients responding to immunotherapy22. Thus, algorithms that better predict MHC-II neoepitopes would be valuable tools to accelerate validation of neoantigens.
In sum, many factors that contribute to the selection of a therapeutically effective neoepitope are outside the scope of the predictions enabled by in silico approaches. Arguably, the rules of binding and immunogenicity currently used were established in viral systems analyzing thousands of peptides. Similar large-scale studies on tumor mutant peptides have just begun5. Moreover, whereas the operational criteria for viral peptides reflect situations not influenced by the chronicity of the disease, tumor neoepitopes exist in the context of a chronic, evolving disease associated with immune suppression. Not surprisingly, it has been shown that the repertoire for tumor neoepitopes is larger in the peripheral blood of naive individuals than among the T cells infiltrating a tumor23. Experimental validation of immunogenicity24 is, therefore, a crucial step in improving the odds of successful immunotherapy. Ideally, immunogenicity should be established using a combination of in vitro stimulation of peripheral blood T cells from normal donors matched to the HLA of the patient and/or in vivo immunization of human leukocyte antigen (HLA)-transgenic mice23,25,26. This combined approach has provided clear-cut results in viral systems and should be used to validate tumor neoepitopes in each patient. This was shown to be applicable to mutant tumor antigen peptides27, even though the T cell repertoire of HLA transgenic mice may be wider than that of vaccinated human individuals with cancer28. If the peptide is immunogenic in this context, these TCRs can be engineered onto the patient's T cells and reintroduced as an adoptive T-cell therapy. Until very recently the guiding principle of therapeutic cancer vaccines was to use conserved tumor antigens. This off-the shelf approach, however convenient, proved disappointing. Now, new approaches leveraging genomic and informatics tools to rapidly identify mutant peptides fall short of identifying truly immunogenic peptides. Thus, sacrificing time to improve efficiency seems unavoidable, considering that presently only a fraction of predicted mutant peptides are immunogenic. Perhaps, attention should be turned to developing new fast assay systems to better validate immunogenicity.
The advantages of a thorough validation are clear: to avoid formulating immunogens comprising multiple peptides where only a fraction is demonstrably immunogenic and recognize the cancer cell. Narrowing neoepitopes to those truly immunogenic avoids raising T-cell immunity against antigens of unknown function limiting the risk of adverse effects but also increasing the efficacy of vaccination since the few available studies show that only a fraction (<30%) of neoepitopes selected either through MHC binding or MS are immunogenic5,11. Importantly, immunogenic neoepitopes should not include peptides that are not processed in the tumor cell (cryptic) as the latter are a source of intrapeptide competition in the antigen-presenting cell, hindering the anti-tumor response29,30. Based on the foregoing, we advocate that resources be devoted to develop high-throughput methods that allow a more precise and rapid validation of predicted immunogenic neoepitopes for clinical use.
Of course, there is no guarantee of clinical efficacy, even with validated neoepitopes. To date, clinical efficacy of cancer vaccination has been documented sporadically and more data are needed. In the interim, research on personalized genomic immunotherapy would benefit greatly from more extensive pre-publication sharing of negative findings with immunized patients. Such data may well hold the answers to some of the questions raised here.
Note added in proof: Recently Ott et al. reported on the immunization of six patients with advanced melanoma with 97 neopeptides (Ott, P.A. et al., Nature 547, 217-221 (2017)). A careful study showed that CD4 and CD8 T cell responses were elicited to a substantial number of these neoepitopes (60% for CD4 and 16% for CD8 T cells), although only a small subset of neoepitope-specific T cell lines (three) could be shown to recognize autologous melanoma cells. It would be of interest to know how the 97 neopeptides identified in this study would fare against the validation scheme proposed herein.
References
- Boon, T., Cerottini, J.C., Van den Eynde, B., van der Bruggen, P. & Van Pel, A. Annu. Rev. Immunol. 12, 337–365 (1994).
Article CAS Google Scholar - Snyder, A. et al. N. Engl. J. Med. 371, 2189–2199 (2014).
Article Google Scholar - Rizvi, N.A. et al. Science 348, 124–128 (2015).
Article CAS Google Scholar - Van Allen, E.M. et al. Science 350, 207–211 (2015).
Article CAS Google Scholar - Bassani-Sternberg, M. et al. Nat. Commun. 7, 13404 (2016).
Article CAS Google Scholar - Gfeller, D., Bassani-Sternberg, M., Schmidt, J. & Luescher, I.F. Oncoimmunology 5, e1177691 (2016).
Article Google Scholar - Sykulev, Y., Joo, M., Vturina, I., Tsomides, T.J. & Eisen, H.N. Immunity 4, 565–571 (1996).
Article CAS Google Scholar - Calis, J.J. et al. PLoS Comput. Biol. 9, e1003266 (2013).
Article Google Scholar - Paul, S. et al. J. Immunol. 191, 5831–5839 (2013).
Article CAS Google Scholar - Yewdell, J.W. Immunity 25, 533–543 (2006).
Article CAS Google Scholar - Carreno, B.M. et al. Science 348, 803–808 (2015).
Article CAS Google Scholar - Sette, A. et al. J. Immunol. 153, 5586–5592 (1994).
CAS PubMed Google Scholar - Akram, A. & Inman, R.D. Eur. J. Immunol. 43, 3254–3267 (2013).
Article CAS Google Scholar - Sidney, J., Peters, B., Frahm, N., Brander, C. & Sette, A. BMC Immunol. 9, 1 (2008).
Article Google Scholar - Abelin, J.G. et al. Immunity 46, 315–326 (2017).
Article CAS Google Scholar - McGranahan, N. et al. Science 351, 1463–1469 (2016).
Article CAS Google Scholar - Alexandrov, L.B. et al. Nature 500, 415–421 (2013).
Article CAS Google Scholar - Klein, L., Kyewski, B., Allen, P.M. & Hogquist, K.A. Nat. Rev. Immunol. 14, 377–391 (2014).
Article CAS Google Scholar - Arstila, T.P. et al. Science 286, 958–961 (1999).
Article CAS Google Scholar - Brodin, P. et al. Cell 160, 37–47 (2015).
Article CAS Google Scholar - Zanetti, M. J. Immunol. 194, 2049–2056 (2015).
Article CAS Google Scholar - Spitzer, M.H. et al. Cell 168, 487–502.e15 (2017).
Article CAS Google Scholar - Strønen, E. et al. Science 352, 1337–1341 (2016).
Article Google Scholar - Vitiello, A. et al. J. Immunol. 157, 5555–5562 (1996).
CAS PubMed Google Scholar - Vitiello, A., Marchesini, D., Furze, J., Sherman, L.A. & Chesnut, R.W. J. Exp. Med. 173, 1007–1015 (1991).
Article CAS Google Scholar - Wang, Q.J. et al. Cancer Immunol. Res. 4, 204–214 (2015).
Article Google Scholar - Hernandez, J. et al. Proc. Natl. Acad. Sci. USA 99, 12275–12280 (2002).
Article CAS Google Scholar - Schmidt, J. et al. J. Biol. Chem. 292, 11840–11849 (2017).
Article CAS Google Scholar - Gnjatic, S. et al. Proc. Natl. Acad. Sci. USA 99, 11813–11818 (2002).
Article CAS Google Scholar - Willis, R.A., Kappler, J.W. & Marrack, P.C. Proc. Natl. Acad. Sci. USA 103, 12063–12068 (2006).
Article CAS Google Scholar
Author information
Authors and Affiliations
- PersImmune, Inc., San Diego, California, USA
Antonella Vitiello - Department of Medicine and Moores Cancer Center, The Laboratory of Immunology, University of California San Diego, La Jolla, California, USA
Maurizio Zanetti
Authors
- Antonella Vitiello
You can also search for this author inPubMed Google Scholar - Maurizio Zanetti
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toAntonella Vitiello or Maurizio Zanetti.
Ethics declarations
Competing interests
A.V. is the CEO of PersImmune Inc., a biotech company focused on the development of personalized immunotherapy.
Rights and permissions
About this article
Cite this article
Vitiello, A., Zanetti, M. Neoantigen prediction and the need for validation.Nat Biotechnol 35, 815–817 (2017). https://doi.org/10.1038/nbt.3932
- Published: 11 September 2017
- Issue Date: September 2017
- DOI: https://doi.org/10.1038/nbt.3932