Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder (original) (raw)
- Letter
- Published: 01 August 2004
- Elisa Romeo3,
- Zorica Ristic3,
- Toshihiro Ohura4,
- Caroline Stuart1,
- Mauricio Arcos-Burgos1,
- Mital H Dave3,
- Carsten A Wagner3,
- Simone R M Camargo3,
- Sumiko Inoue5,
- Norio Matsuura5,
- Amanda Helip-Wooley1,
- Detlef Bockenhauer6,
- Richard Warth7,
- Isa Bernardini1,
- Gepke Visser8,
- Thomas Eggermann9,
- Philip Lee10,
- Arthit Chairoungdua11,
- Promsuk Jutabha11,
- Ellappan Babu11,
- Sirinun Nilwarangkoon11,
- Naohiko Anzai11,
- Yoshikatsu Kanai11 na1,
- Francois Verrey3 na1,
- William A Gahl1,2 na1 &
- …
- Akio Koizumi5 na1
Nature Genetics volume 36, pages 999–1002 (2004)Cite this article
- 6687 Accesses
- 234 Citations
- 3 Altmetric
- Metrics details
Abstract
Hartnup disorder, an autosomal recessive defect named after an English family described in 1956 (ref. 1), results from impaired transport of neutral amino acids across epithelial cells in renal proximal tubules and intestinal mucosa. Symptoms include transient manifestations of pellagra (rashes), cerebellar ataxia and psychosis1,2. Using homozygosity mapping in the original family in whom Hartnup disorder was discovered, we confirmed that the critical region for one causative gene was located on chromosome 5p15 (ref. 3). This region is homologous to the area of mouse chromosome 13 that encodes the sodium-dependent amino acid transporter B0AT1 (ref. 4). We isolated the human homolog of B0AT1, called SLC6A19, and determined its size and molecular organization. We then identified mutations in SLC6A19 in members of the original family in whom Hartnup disorder was discovered and of three Japanese families. The protein product of SLC6A19, the Hartnup transporter, is expressed primarily in intestine and renal proximal tubule and functions as a neutral amino acid transporter.
Similar content being viewed by others
Main
Despite molecular characterization of other proximal tubule transporters, the neutral amino acid carrier defective in Hartnup disorder (OMIM 2345000) has resisted genetic identification2. We carried out homozygosity mapping and fine mapping in ten members of two consanguineous families (the siblings in whom Hartnup disorder was originally discovered1; family A; Fig. 1a) and in siblings from the US5 (family B; Fig. 1a). We found linkage of Hartnup disorder to 5p15 only in family A, with a maximum combined multipoint lod score of 2.31 at 11.24 cM (P = 0.01). This confirmed our previous results showing linkage to chromosome 5p15 (ref. 3). In family B, we obtained a maximum multipoint lod score of −2.40 at 15.81 cM.
Figure 1: Families with Hartnup disorder and topology of B0AT1 and its mutations.
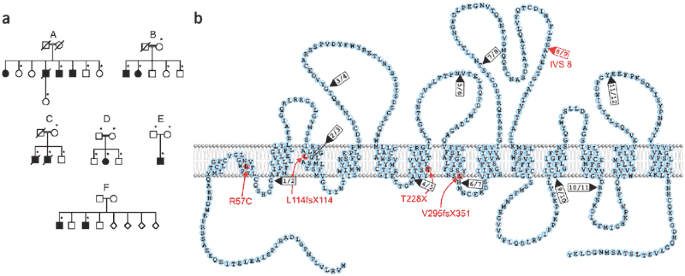
(a) Families with Hartnup disorder. In family A (in whom Hartnup disorder was originally discovered), a splice site mutation in SLC6A19 segregates with Hartnup disorder. Asterisks indicate individuals from whom DNA was available for analysis. (b) Depiction of B0AT1 in the plasma membrane. Mutations are indicated in red; arrowheads indicate splice sites.
We simultaneously pursued two mouse monoamine transporter-related orphan genes, Slc6a18 (also called Xtrp2; ref. 6) and Slc6a19 (encoding B0AT1; ref. 4). These members of the SLC6 family of transporters map to the mouse chromosomal region that is homologous to human chromosome 5p15. Both Slc6a18 and Slc6a19 showed abundant expression in mouse kidney, as assessed by real time RT-PCR (Fig. 2a). Immunohistochemistry confirmed expression of mouse B0AT1 at the brush border of small intestine (data not shown) and kidney proximal tubule cells (Fig. 2b).
Figure 2: Expression of B0AT1 and Slc6a18 in mouse tissues and human tissue expression profile for the Hartnup transporter SLC6A19 (encoding B0AT1).
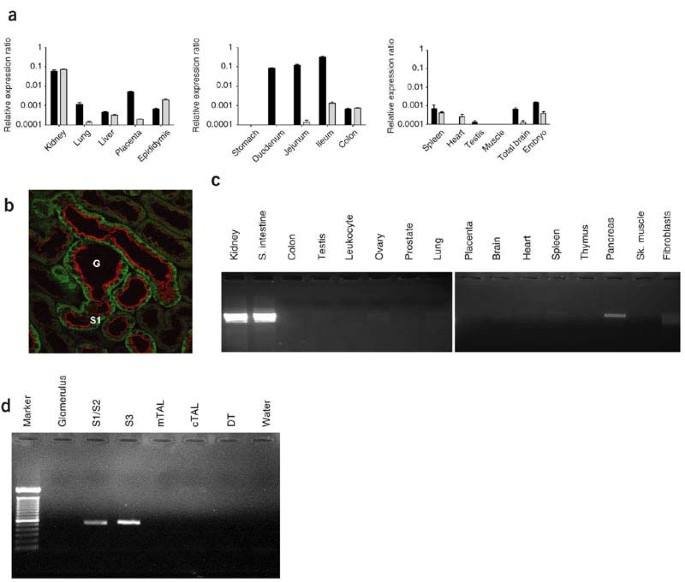
(a) Relative expression ratios from real-time RT-PCR expression profiles of mouse Slc6a19 (black bars) and mouse Slc6a18 (gray bars). Error bars represent s.e.m. (b) Immunofluorescence of B0AT1 (red) and of the basolateral protein 4F2hc (green) indicates the luminal expression of B0AT1 in kidney proximal tubule (highest in segment S1). G, glomerulus. The proximal tubule cells of mouse kidney start in the glomerular capsule. (c) PCR amplification of SLC6A19 cDNA in a panel of human tissues. SLC6A19 was highly expressed in kidney and small intestine, and weakly expressed in pancreas. (d) SLC6A19 cDNA was amplified from cells isolated from all segments of human proximal tubule (S1/S2 and S3), but not from the glomerulus, medullary thick ascending limb (mTAL), cortical thick ascending limb (cTAL) or distal tubule (DT).
The human homolog, B0AT1, is encoded by the predicted locus SLC6A19, with a 2,022-bp open reading frame. PCR amplification using human kidney cDNA produced a 1,905-bp product with 100% identity to SLC6A19 sequence. We next determined the genomic organization of SLC6A19, which has a stop codon 28 bases before the ATG in the 5′ untranslated region. SLC6A19 has 12 coding exons. The B0AT1 protein contains 634 amino acids and 12 predicted transmembrane regions (Fig. 1b). In a panel of human cDNAs, we detected robust expression of SLC6A19 in kidney and small intestine, with minimal expression in pancreas (Fig. 2c). SLC6A19 was also expressed in stomach, liver, duodenum and ileocecum (data not shown). In contrast, human SLC6A18 was abundantly expressed in human kidney but not in human intestine (data not shown). These findings indicated that SLC6A19 was the primary candidate for the gene causing Hartnup disorder.
We sequenced SLC6A19 in six families with typical neutral aminoaciduria (Fig. 1a) and identified five mutations in SLC6A19 (deletion, nonsense, missense and splice site) in four families (Table 1). Family A had a homozygous splice site mutation, IVS8 + 2T → G, that segregated with the phenotype. Family C (Japanese) had two affected individuals who were homozygous with respect to 884–885delTG in exon 3. A single affected member of family D (Japanese) had a homozygous missense mutation in exon 1, 169C → T, changing the conserved Arg57 to a cysteine residue. This occurred in the first membrane segment of B0AT1 (Fig. 1b) and was not present in 100 Japanese control alleles. We identified an individual with asymptomatic Hartnup disorder in family E (Japanese; Fig. 1a) by neonatal mass screening. He was compound heterozygous with respect to c340delC in exon 2 and the nonsense mutation 682–683AC → TA in exon 5, with termination at amino acid 228. Families B and F, American sibships with neutral aminoaciduria5,7, lacked mutations in exons 1–12 of SLC6A19 and in the gene's splice sites. This finding, and the absence of linkage to chromosome 5p15 in family B, suggests that there is locus heterogeneity. Other genes, such as those encoding a complementary transporter protein, a putative associated protein, a subunit or an efflux transporter at the basolateral side of proximal tubule cells, may be involved in transepithelial neutral amino acid transport. These possibilities have precedent in the proximal tubule transport of dibasic amino acids, which is impaired in cystinuria by defects in either subunit of the heterodimeric apical cystine transporter rBAT-b0,+AT or, in the case of lysinuric protein intolerance, in the basolateral transporter 4F2hc-y+LAT1 (refs. 8,9).
Table 1 Mutations in SLC6A19 associated with Hartnup disorder
The cellular expression of SLC6A19 was consistent with its putative role in plasma membrane transport. We amplified total cDNA from different portions of human renal tubules by PCR using SLC6A19 primers. Expression was apparent in all segments of the proximal tubules, but not in the glomerulus, thick ascending limb or distal tubule (Fig. 2d). Mouse B0AT1 transported neutral amino acids in Xenopus laevis oocytes; the Km for leucine was ∼0.63 mM, and transport was sodium-dependent and specific for neutral amino acids4. We expressed human B0AT1 in oocytes and found sodium-dependent neutral amino acid transport with a Km for leucine of 1.2 mM (Fig. 3a–c). Transport was abolished by the missense mutation R57C in family D (Fig. 3d).
Figure 3: Function of SLC6A19 (encoding B0AT1).
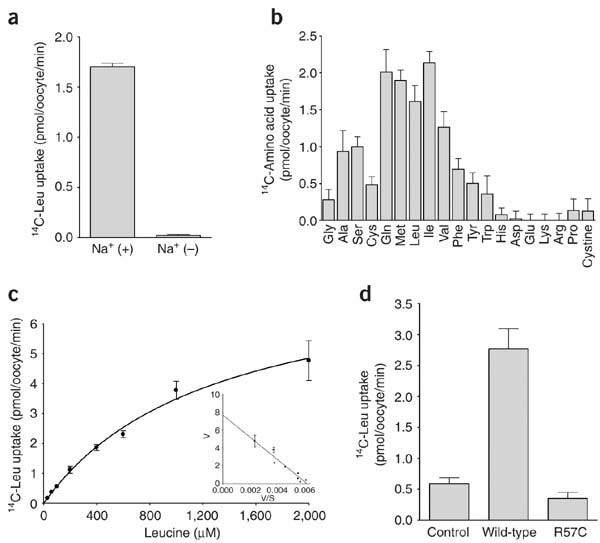
(a) B0AT1-mediated transport of L-leucine (1 mM) was sodium-dependent. (b) The transport of 14C-labeled amino acids (1 mM) mediated by _h_B0AT1 demonstrated substrate selectivity for neutral amino acids. (c) B0AT1-mediated transport of 14C-labeled leucine was saturable and conformed to Michaelis-Menten kinetics, with Km of 1.2 mM and Vmax of 7.7 pmol per oocyte per min. Inset, Eadie-Hofstee plot of leucine uptake. (d) The R57C mutant of B0AT1 (found in family D) yielded minimal uptake of 14C-labeled leucine (1 mM). Error bars represent s.e.m.
On the basis of mapping data, tissue and cellular expression, functional studies, and mutation identification in the family in whom Hartnup disorder was originally discovered and in three other families, we conclude that B0AT1 is the Hartnup transporter, joining the transporters in the plasma membrane that are involved in human disease10. Similar results and conclusions were found in Australian individuals with Hartnup disorder11.
Methods
Mapping.
We obtained genomic DNA from whole blood or as published previously3 from members of families A, B, C, D and E after obtaining informed consent under a protocol approved by the National Human Genome Research Institute Institutional Review Board3. For family F, we extracted DNA from cells obtained from the Coriell Human Mutant Cell Repository. A genome scan using automated fluorescent microsatellite analysis of 2,011 markers, with an average marker density of 2 cM throughout the genome and an average marker heterozygosity of ∼0.75, was done at deCODE Genetics. For fine mapping between D5S1981 and D5S464, we used 11 polymorphic markers with an average spacing of 426 kb. We carried out linkage analyses using mapping by homozygosity with the software HOMOZ that calculates multipoint lod scores in pedigrees with inbreeding loops. We carried out two-point and multipoint parametric and nonparametric analyses of linkage using LINKAGE, with the speed improvement implemented in FASTLINK and GENEHUNTER, respectively12,13,14. We used the high-performance computational capabilities of the SGI Origin 2000 system at the Center for Information Technology of the National Institutes of Health.
Sequencing and mutation analysis.
We chose intronic primers (Invitrogen) to sequence exons and splice sites of each exon. Primer sequences are available on request. We separated the PCR products of each exon by electrophoresis on agarose gels and removed specific bands for DNA isolation. We sequenced this DNA in both directions using a Beckman Coulter CEQ8000 following the manufacturer's protocol or as published previously3.
RNA extraction and reverse transcription.
We killed male C57BL6 mice by intraperitoneal injection of ketamine and xylazine followed by cervical dislocation. We then collected tissues and rapidly froze them until further use. We collected stomach, duodenum, jejunum, ileum and colon, rinsed them several times with ice-cold phosphate buffered saline (PBS; pH 7.4), scraped off the mucosa cell layers on ice and rapidly froze them. For total RNA extraction, we thawed tissues in RLT buffer of the RNeasy Mini Kit (Qiagen) containing 10 μl ml−1 of β-mercaptoethanol (Sigma) and homogenized them on ice. RNA was bound to columns and treated with DNase for 15 min at 25 °C to reduce genomic DNA contamination. We used 100 ng of RNA as template for reverse transcription with TaqMan Reverse Transcription Kit (Applied Biosystems) in the presence of 2.5 μM of random hexamer primers.
Real-time PCR.
We carried out real-time PCR using cDNA template and TaqMan Universal PCR master mix (Applied Biosystems). We chose primers to result in amplicons of 70–100 bp that span intron-exon boundaries. We labeled probes with reporter dye FAM at the 5′ end and the quencher dye TAMRA at the 3′ end (Microsynth). Reactions were done in 96-well optical reaction plates using a Prism 7700 cycler (Applied Biosystems). After 10 min at 95 °C, we carried out 40 cycles of 95 °C for 15 s and 60 °C for 1 min with auto-ramp time. For data analysis, we set the threshold to 0.06 (value in the linear range of amplification curves). All reactions were run in duplicate. We calculated the abundance of target mRNA relative to a reference mRNA (GAPD). Assuming an efficiency of 2 (relative increase in template mRNA required to decrease by 1 the number of cycles), we calculated relative expression ratios as R = 2(Ct(GAPD)−Ct(test)), where Ct is the cycle number at the threshold and test is the tested mRNAs.
Immunohistochemistry.
We anesthetized male C57BL6 mice (10–12 weeks old) with ketamine and xylazine and perfused them through the left ventricle with PBS followed by a buffered paraformaldehyde solution (4%, pH 7). We removed kidneys and small intestine, flushed them with fixative solution, cut them and embedded them in Tissue-Tek V.I.P. medium (Sakura) just before freezing in liquid nitrogen. We cut 5-μm serial sections with a cryostat and collected them on polylysine-coated slides. We incubated sections for 5 min in 0.1% SDS, washed them three times and then incubated them with the primary antibody (diluted in PBS, 0.04% Triton X-100) for 1 h at room temperature. We again washed sections three times and then incubated them with the secondary antibody (diluted in PBS, 0.04% Triton X-100) for 1 h at room temperature. After washing them five times, we mounted the sections using DAKO mounting medium and viewed them with a Leica SP1 UV CLSM confocal microscope. We used rabbit antibody to mouse B0AT1 (antigen peptide NH2-MVRLVLPNPGLEERIC-CONH2; serum, dilution 1:200) and goat antibody to mouse 4F2hc (Santa Cruz Biotech, 1:400) as primary antibodies and donkey antibody to rabbit (1:1,000) and donkey antibody to goat (1:500; Molecular Probes) as secondary antibodies.
Segmental expression in human kidney.
We carried out segment-specific dissection according to a previously described protocol15. We extracted total RNA and reverse transcribed it to generate segment-specific cDNA as described16.
Functional studies.
We carried out RT-PCR of SLC6A19 cDNA (primer sequences available on request) on the first-strand cDNA generated from human kidney poly(A)+ RNA (Clontech). We cloned the PCR fragment into pCR2.1-TOPO vector (Invitrogen). For the expression in oocytes, we excised the SLC6A19 cDNA with _Eco_RI and subcloned it into pcDNA3.1(+) vector (Invitrogen). We carried out site-directed mutagenesis (R57C) of SLC6A19 cDNA in the pcDNA3.1(+) plasmid using the QuikChange kit (Stratagene). We checked the accuracy of the cDNAs by direct sequencing. We injected X. laevis oocytes with 25 ng of cRNA synthesized in vitro from SLC6A19 cDNA in the pcDNA3.1(+) plasmid linearized with _Not_I. We carried out in vitro transcription using the T7 mMESSAGE mMACHINE Kit (Ambion) and polyadenylation of the cRNA using the Poly(A) Tailing Kit (Ambion). We measured transport after 2–3 d of expression as described17. We measured uptake of 14C-labeled amino acids in the regular uptake solution (100 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES buffer (pH 7.4)) or in a solution in which NaCl was replaced by choline-Cl, containing 2.5 μCi ml−1 of radioactively labeled compounds and 0.03–2 mM of amino acids. We measured uptake (pmol per oocyte per min) in triplicate for 30 min.
GenBank accession numbers.
SLC6A19 open reading frame, XM_291120; SLC6A19 cDNA sequence, AY596807.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
- Baron, D.N., Dent, C.E., Harris, H., Hart, E.W. & Jepson, J.B. Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria, and other bizarre biochemical features. Lancet 271, 421–428 (1956).
Article CAS PubMed Google Scholar - Levy, H.L. Hartnup disorder. in The Metabolic and Molecular Bases of Inherited Disease 8th edn., vol. III (eds. Scriver, C.R., Beaudet, A.L., Valle, D.L. & Sly, W.S.) 4957–4969 (McGraw-Hill, New York, 2001).
Google Scholar - Nozaki, J. et al. Homozygosity mapping to chromosome 5p15 of a gene responsible for Hartnup disorder. Biochem. Biophys. Res. Commun. 284, 255–260 (2001).
Article CAS PubMed Google Scholar - Broer, A. et al. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J. Biol. Chem. 279, 24467–24476 (2004).
Article CAS PubMed Google Scholar - Bernardini, I., Introne, W., Kleta, R., Fitzpatrick, D.L. & Gahl, W.A. Detection of Hartnup's disorder in an alkaptonuria sibship. Am. J. Hum. Genet. 69, Suppl. 1, 1784 (2001).
Google Scholar - Virlon, B. et al. Serial microanalysis of renal transcriptomes. Proc. Natl. Acad. Sci. USA 96, 15286–15291 (1999).
Article CAS PubMed PubMed Central Google Scholar - Shih, V.E., Bixby, E.M., Alpers, D.H., Bartsocas, C.S. & Thier, S.O. Studies of intestinal transport defect in Hartnup disease. Gastroenterology 61, 445–453 (1971).
CAS PubMed Google Scholar - Verrey, F. et al. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 447, 532–542 (2004).
Article CAS PubMed Google Scholar - Palacin, M., Goodyer, P., Nunes, V., Gasparini & Cystinuria, P. in The Metabolic and Molecular Bases of Inherited Disease 8th edn., vol. III (eds. Scriver, C.R., Beaudet, A.L., Valle, D.L. & Sly, W.S.) 4909–4932 (McGraw-Hill, New York, 2001).
Google Scholar - Kleta, R., Stuart, C., Gill, F.A. & Gahl, W.A. Renal glucosuria due to SGLT2 mutations. Mol. Genet. Metab. 82, 56–58 (2004).
Article CAS PubMed Google Scholar - Seow, H.F. et al. Hartnup disorder is caused by mutations in SLC6A19, encoding the neutral amino acid transporter. Nat. Genet. advance online publication, 1 August 2004 (doi:10.1038/ng′).
- Lander, E.S. & Botstein, D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236, 1567–1570 (1987).
Article CAS PubMed Google Scholar - Kruglyak, L., Daly, M.J. & Lander, E.S. Rapid multipoint linkage analysis of recessive traits in nuclear families, including homozygosity mapping. Am. J. Hum. Genet. 56, 519–527 (1995).
CAS PubMed PubMed Central Google Scholar - Cottingham, R.W. Jr., Idury, R.M. & Schaffer, A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet. 53, 252–263 (1993).
PubMed PubMed Central Google Scholar - Schafer, J.A. et al. A simplified method for isolation of large numbers of defined nephron segments. Am. J. Physiol. 273, F650–F657 (1997).
CAS PubMed Google Scholar - Levy, D.I., Velazques, H., Goldstein, S.A. & Bockenhauer, D. Segment-specific expression of 2P domain potassium channel genes in human nephron. Kidney Int. 65, 918–926 (2004).
Article CAS PubMed Google Scholar - Kanai, Y. & Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 360, 467–471 (1992).
Article CAS PubMed Google Scholar
Acknowledgements
We thank D. Bacic for help with frozen sections, L.G. Palacio for statistical analyses, F. Skovby for expert advice and the families who graciously participated in this study. This study was supported by Grants-in-aid from the Japanese Government to A.K. and by Swiss National Science Foundation grants to F.V.
Author information
Author notes
- Yoshikatsu Kanai, Francois Verrey, William A Gahl and Akio Koizumi: These authors contributed equally to this work.
Authors and Affiliations
- Medical Genetics Branch, 10 Center Drive, MSC 1851, Building 10, Room 10C-107, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland, USA
Robert Kleta, Caroline Stuart, Mauricio Arcos-Burgos, Amanda Helip-Wooley, Isa Bernardini & William A Gahl - Office of Rare Diseases, Intramural Program, Office of the Director, National Institutes of Health, Bethesda, Maryland, USA
Robert Kleta & William A Gahl - Institute of Physiology, University of Zurich, Zurich, Switzerland
Elisa Romeo, Zorica Ristic, Mital H Dave, Carsten A Wagner, Simone R M Camargo & Francois Verrey - Department of Pediatrics, Tohoku University School of Medicine, Sendai, Japan
Toshihiro Ohura - Kyoto University Graduate School of Medicine, Kyoto, Japan
Sumiko Inoue, Norio Matsuura & Akio Koizumi - Department of Pediatrics, Yale University School of Medicine, New Haven, Connecticut, USA
Detlef Bockenhauer - Institut fuer Physiologie der Universitaet Regensburg, Regensburg, Germany
Richard Warth - Department of Metabolic Diseases, Wilhelmina Children's Hospital, University Hospital Utrecht, Utrecht, The Netherlands
Gepke Visser - Institute of Human Genetics, Aachen University Hospital, Aachen, Germany
Thomas Eggermann - The Charles Dent Metabolic Unit, The National Hospital for Neurology and Neurosurgery, UK
Philip Lee - Department of Pharmacology and Toxicology, Kyorin University School of Medicine, Mitaka, Tokyo, Japan
Arthit Chairoungdua, Promsuk Jutabha, Ellappan Babu, Sirinun Nilwarangkoon, Naohiko Anzai & Yoshikatsu Kanai
Authors
- Robert Kleta
You can also search for this author inPubMed Google Scholar - Elisa Romeo
You can also search for this author inPubMed Google Scholar - Zorica Ristic
You can also search for this author inPubMed Google Scholar - Toshihiro Ohura
You can also search for this author inPubMed Google Scholar - Caroline Stuart
You can also search for this author inPubMed Google Scholar - Mauricio Arcos-Burgos
You can also search for this author inPubMed Google Scholar - Mital H Dave
You can also search for this author inPubMed Google Scholar - Carsten A Wagner
You can also search for this author inPubMed Google Scholar - Simone R M Camargo
You can also search for this author inPubMed Google Scholar - Sumiko Inoue
You can also search for this author inPubMed Google Scholar - Norio Matsuura
You can also search for this author inPubMed Google Scholar - Amanda Helip-Wooley
You can also search for this author inPubMed Google Scholar - Detlef Bockenhauer
You can also search for this author inPubMed Google Scholar - Richard Warth
You can also search for this author inPubMed Google Scholar - Isa Bernardini
You can also search for this author inPubMed Google Scholar - Gepke Visser
You can also search for this author inPubMed Google Scholar - Thomas Eggermann
You can also search for this author inPubMed Google Scholar - Philip Lee
You can also search for this author inPubMed Google Scholar - Arthit Chairoungdua
You can also search for this author inPubMed Google Scholar - Promsuk Jutabha
You can also search for this author inPubMed Google Scholar - Ellappan Babu
You can also search for this author inPubMed Google Scholar - Sirinun Nilwarangkoon
You can also search for this author inPubMed Google Scholar - Naohiko Anzai
You can also search for this author inPubMed Google Scholar - Yoshikatsu Kanai
You can also search for this author inPubMed Google Scholar - Francois Verrey
You can also search for this author inPubMed Google Scholar - William A Gahl
You can also search for this author inPubMed Google Scholar - Akio Koizumi
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRobert Kleta.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Kleta, R., Romeo, E., Ristic, Z. et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder.Nat Genet 36, 999–1002 (2004). https://doi.org/10.1038/ng1405
- Received: 20 April 2004
- Accepted: 17 June 2004
- Published: 01 August 2004
- Issue Date: 01 September 2004
- DOI: https://doi.org/10.1038/ng1405