Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer (original) (raw)
- Letter
- Published: 08 August 2004
- Damaris Ponciano-Jackson2,
- Maija Wolf3,
- Jeff A Kiefer1,
- David O Azorsa1,
- Sukru Tuzmen1,
- Don Weaver1,
- Christiane Robbins2,
- Tracy Moses2,
- Minna Allinen4,
- Sampsa Hautaniemi5,
- Yidong Chen4,
- Abdel Elkahloun4,
- Mark Basik6,
- G Steven Bova7,
- Lukas Bubendorf8,
- Alessandro Lugli8,
- Guido Sauter8,
- Johanna Schleutker9,
- Hilmi Ozcelik10,
- Sabine Elowe10,
- Tony Pawson10,
- Jeffrey M Trent2,
- John D Carpten2,
- Olli-P Kallioniemi1,3 &
- …
- Spyro Mousses1
Nature Genetics volume 36, pages 979–983 (2004)Cite this article
- 2877 Accesses
- 4 Altmetric
- Metrics details
Abstract
The identification of tumor-suppressor genes in solid tumors by classical cancer genetics methods is difficult and slow. We combined nonsense-mediated RNA decay microarrays1 and array-based comparative genomic hybridization2,3 for the genome-wide identification of genes with biallelic inactivation involving nonsense mutations and loss of the wild-type allele. This approach enabled us to identify previously unknown mutations in the receptor tyrosine kinase gene EPHB2. The DU 145 prostate cancer cell line, originating from a brain metastasis, carries a truncating mutation of EPHB2 and a deletion of the remaining allele. Additional frameshift, splice site, missense and nonsense mutations are present in clinical prostate cancer samples. Transfection of DU 145 cells, which lack functional EphB2, with wild-type EPHB2 suppresses clonogenic growth. Taken together with studies indicating that EphB2 may have an essential role in cell migration and maintenance of normal tissue architecture, our findings suggest that mutational inactivation of EPHB2 may be important in the progression and metastasis of prostate cancer.
Similar content being viewed by others
Main
Inactivation of tumor-suppressor genes (TSGs) in cancer is often a two-step process4 involving mutation of the target gene and loss of the wild-type allele. Mapping of chromosomal deletions and losses of heterozygosity in cancer cells has been widely applied to guide the identification of TSGs. On its own, however, this approach is slow, labor-intensive and complicated by genomic instability, which often leads to numerous candidate regions for further study. In an alternative approach, the nonsense-mediated decay (NMD) mechanism, which normally targets transcripts with nonsense mutations for rapid degradation5,6, is blocked to cause the differential stabilization of genes that contain truncating mutations. This approach, coupled with microarrays to measure transcript levels after NMD inhibition, has been proposed for the genome-wide identification of mutated genes in cell lines1.
Here we combined results from NMD microarray experiments highlighting putative nonsense mutations with high-resolution data on deleted genomic regions in cancer cell lines obtained with array-based comparative genomic hybridization (CGH)2,3. We applied this integrated approach, which focuses on biallelic gene inactivation events, to the identification of candidate TSGs in prostate cancer.
We pretreated the DU 145, PC-3 and LNCaP prostate cancer cell lines with emetine (which inhibits the NMD pathway) and then exposed them to actinomycin D to block new mRNA synthesis and to distinguish post-transcriptional shifts in mRNA stability, which indicate the presence of a nonsense mutation. We used cDNA microarrays to measure changes in transcript levels in cells treated with emetine versus untreated cells. We also carried out corresponding analyses with nonmalignant control cells to distinguish drug-induced gene expression changes from mutation-induced transcript stabilization events. We used known nonsense mutations, including the C39X amino acid mutation in MLH1 in DU 145 cells and the A138X mutation in TP53 in PC-3 cells, as positive controls to optimize the technology and validate the strategy.
As predicted, the normalized NMD microarray ratios for both MLH1 and TP53 were elevated specifically in the cell lines carrying truncating mutations in these genes, but not in the cell lines lacking such mutations. We validated the accumulation of these mutated transcripts by quantitative RT-PCR (Fig. 1). In addition, cDNA microarray-based CGH indicated that the TP53 locus was deleted in PC-3 cells and the MLH1 locus was deleted in DU 145 cancer cells (Supplementary Fig. 1 online).
Figure 1: Relative changes in transcript levels resulting from emetine-mediated NMD blockade.
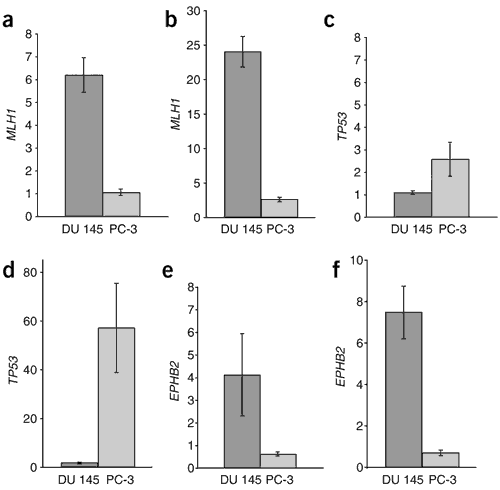
DU 145 and PC-3 cell lines were pretreated with emetine to block the NMD pathway and then treated with actinomycin D to block new transcription. Transcripts were detected and measured by cDNA microarray (a,c,e) and validated by quantitative RT-PCR analysis using gene-specific primers (b,d,f). Both sets of data represent relative expression ratios (emetine-treated versus untreated) averaged across the time course. Error bars indicate standard error between the time points. Data are shown for MLH1 (a,b), TP53 (c,d) and EPHB2 (e,f). An increase in relative transcript levels was observed only in cell lines with known (MLH1 in DU 145, TP53 in PC-3) or new (EPH2B in DU 145) nonsense mutations. Differences between emetine-treated and untreated samples for all mutations in both microarray and quantitative RT-PCR experiments were significant (P < 0.01), as measured by both the parametric _t_-test (two-tailed) and the nonparametric Wilcoxon rank sum test.
Having validated the approach, we used it to identify candidate genes in three prostate cancer cell lines (DU 145, PC-3 and LNCaP). In these lines, the normalized NMD ratios of 0.4% of the genes on the array (45 in DU 145, 65 in PC-3 and 68 in LNCaP) increased by a factor of at least three. We prioritized these genes by selecting candidates located in deleted regions of the genome in the corresponding cell line. This resulted in 36 prioritized candidate genes (9 in DU 145, 10 in PC-3 and 17 in LNCaP) for analysis by DNA sequencing to detect putative mutations in each cell line (sequencing analysis is still in progress).
One of the first candidates selected for mutation analysis was EPHB2, whose normalized NMD microarray ratio increased in DU 145 cells but not in PC-3 cells (Fig. 1). This increase suggested that post-transcriptional stabilization of mutated EPHB2 mRNA occurred after NMD blockade in DU 145 cells. We confirmed the differential expression of EPHB2 by quantitative RT-PCR (Fig. 1). EPHB2 also mapped to a deleted region at 1p36 by CGH microarray data, further suggesting that it is biallelically inactivated in DU 145 cells (Fig. 2). Notably, this chromosomal locus has been linked to hereditary prostate cancer7. Sequencing confirmed that the DU 145 cell line has a hemizygous nonsense mutation 2167C → T (Table 1 and Supplementary Fig. 2 online) that results in the amino acid change Q723X, truncating EphB2 at the kinase domain (Fig. 3), and is predicted to lead to a complete loss of receptor signaling.
Figure 2: Integration of microarray data from CGH and NMD blockade analysis for genome-wide prioritization of the candidate TSGs.
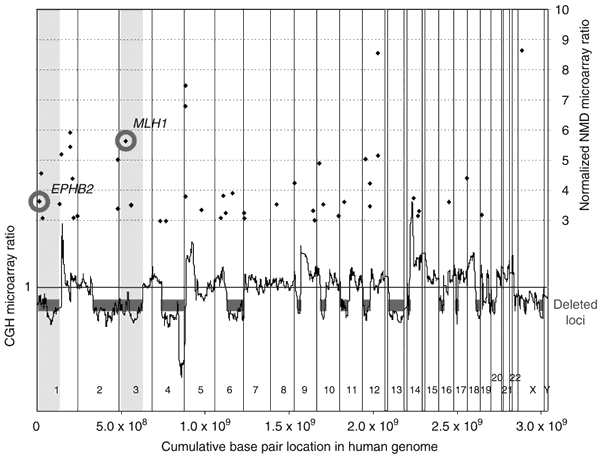
The cumulative base pair location for the whole human genome beginning with 1pter (x axis) was used to map each gene and its copy number status from the CGH microarray analysis of DU 145 cells (black line in bottom half), as well as the corresponding NMD microarray data for genes with an increased NMD ratio in DU 145 cells (black diamonds in top half). The CGH plot was generated by using a moving average of the mean ratios of 30 consecutive clones. Deleted regions are indicated (horizontal gray bar). Genes with a positive mean normalized NMD ratio above 3 are mapped and plotted to their corresponding location. Genes with a positive NMD ratio mapping within deleted loci, such as MLH1 and EPHB2, were prioritized. Vertical gray bars indicate deleted regions corresponding to the EPHB2 and MLH1 loci. Similar results were obtained for PC-3, in which a truncating mutation in TP53 was associated with a positive NMD microarray ratio and mapped to a deleted region of 17p in PC-3 (Supplementary Fig. 1 online).
Table 1 Mutations in EPHB2
Figure 3: Localization of mutations in the EphB2 protein.
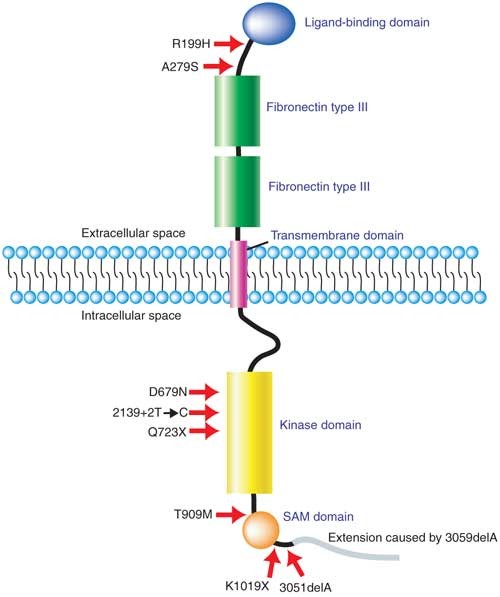
The principal physical structures and domains of EphB2 are shown, and the location of key mutations found in prostate cancer are indicated. Two of the mutations occur in the extracellular part of the protein and six in the intracellular part. The kinase domain, which is essential for receptor signaling, is the most frequently targeted region for mutations. The frameshift mutation 3051delA extends the protein by 72 amino acids, as indicated.
To determine the relevance of this finding to clinical disease, we screened DNA samples obtained from uncultured, clinical prostate tumors, including 33 primary and 62 metastasis specimens, and identified several mutations in EPHB2 (Table 1, Supplementary Table 1 online and Supplementary Fig. 2 online). The most notable was a frameshift mutation, 3051delA, found in three independent metastatic lesions (subdural, humerus and sternum) from the same individual. The mutation was not present in a normal liver sample from this individual, suggesting that it was a somatic mutation. This mutation alters the reading frame at the end of isoform 1 of EPHB2, resulting in an extension of the protein by 72 amino acids. We also found a noncoding mutation in a prostate cancer bone metastasis. This splice site mutation (2139 + 2T → C) destroys the consensus splice donor (GT) of exon 11, which is 100% conserved in eukaryotes. We also observed four previously unknown missense mutations (Table 1 and Fig. 3).
Because we found cancer-associated mutations in EPHB2, we considered that EphB2 might have tumor-suppressor activity. To test this hypothesis, we transfected DU 145 cells (which do not express functional EphB2) with wild-type EphB2 constructs and measured cell growth using colony formation assays. Two human and one mouse wild-type EphB2 expression constructs each suppressed the growth and colony formation of DU 145 cells. The suppression was as efficient as that seen after transfection with wild-type p53, a tumor suppressor that is inactivated by two missense mutations in these cells (Fig. 4). These findings further support the functional relevance of EPHB2 mutations in prostate cancer progression.
Figure 4: EphB2 expression suppresses the growth of DU 145 prostate cancer cells.
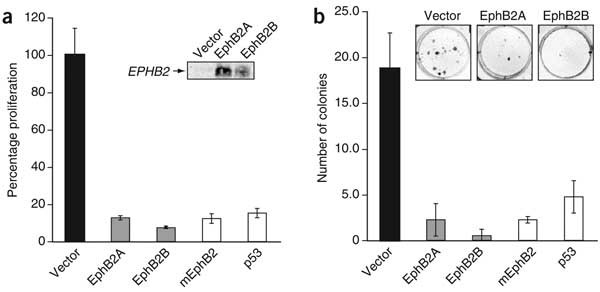
DU 145 cells, with a single truncated form of EphB2, were transfected with control vector or with constructs expressing two human wild-type EphB2 forms (A and B, representing independent subclones), mouse wild-type EphB2 (mEphB2) or wild-type p53. (a) Cell growth was measured with Cell Titer Blue and is shown as a percentage of the growth of cells transfected with control vector. Expression of EphB2 protein was verified by western blotting (inset). (b) All EphB2 expression constructs reduced the growth of DU 145 cells and the number of colonies formed as efficiently as did an expression construct of p53 (which carries the missense mutations P223L and V274F in DU 145).
Eph receptors make up the largest family of receptor tyrosine kinases and can mediate bidirectional signaling through their membrane-associated ephrin ligands. Eph receptors regulate intracellular signaling pathways involved in cell growth, migration, adhesion and polarity. Eph receptors and ephrins are also frequently expressed in complementary patterns that correlate with cellular boundaries during embryonic development8. Consistent with this expression pattern, Eph-ephrin signaling prevents the inappropriate intermingling of distinct cells in culture9 and is required for vascular modeling10,11, axon guidance12,13 and epithelial-mesenchymal transitions14. EphB2 and EphB3 also control the correct positioning of cells in both the embryonic intestinal epithelium and the intestinal crypts.
Disruption of the mouse genes Ephb2 and Ephb3 interferes with normal cellular organization in the crypts, resulting in a loss of normal cell positioning and aberrant mixing of different cell types15. Because loss of cellular borders and disruption of normal tissue architecture are hallmarks of cancer, disruption of EphB2 function may provide a possible molecular mechanism that promotes the spread of cancer cells into normally restricted regions. Eph receptors can also inhibit Ras–mitogen-activated protein kinase signaling16, and their inactivation could therefore contribute to the mitogenic activity of this pathway. This finding is supported by our observation that wild-type EphB2 suppressed clonogenic growth.
Various genetic alterations have been observed throughout the unstable genomes of cancer cells, and these alterations tend to accumulate in advanced stages of disease. The observation of recurrent sequence mutations in the same gene in multiple clinical samples is, however, rare. Multiple recurrent mutations, especially if they are inactivating in nature and associated with loss of heterozygosity, indicate disease-associated changes that accumulate because they confer a selective growth advantage to neoplastic cells. Our genetic and functional data provide evidence that mutations in EPHB2 have a pathogenic role in prostate cancer.
We described an integrated strategy for the genome-wide screening of inactivating mutations in cancer, which has led to the identification of deleterious mutations of EPHB2 in a prostate cancer cell line. Subsequent investigation of clinical samples verified the mutational inactivation of EPHB2 in about 8% of primary and metastatic prostate cancers. We also showed that the expression of wild-type EphB2 in DU 145 cells suppresses their growth. These observations support the idea that EphB2 is a candidate tumor suppressor involved in the progression of prostate cancer. EphB2 is known to be involved in several essential processes, and its disruption may influence neoplastic cell behavior through the loss of intercellular signaling and tissue architecture. Taken together, our findings suggest that loss of EphB2 signaling through mutational inactivation may affect many phenotypic aspects of prostate cancer, such as aberrant growth, invasion and metastasis.
Methods
Cell lines.
We obtained the DU 145, LNCaP and PC-3 prostate cancer cell lines from American Type Culture Collection. We obtained the three nonmalignant cell lines used for normalization of the NMD microarray data (GM11496, GM00156 and GM00038) from Coriell Cell Repository. We grew all cells in accordance with the distributors' instructions in 175-cm2 flasks (Corning) and incubated them at 37 °C.
Clinical specimens.
We obtained 5 primary tumors and 39 metastases from Johns Hopkins University and 28 primary prostate tumors and 23 metastases from the University of Basel. We used a set of 450 normal controls from anonymous blood donors that is commercially available (Coriell). All clinical specimens used in these experiments were from individuals recruited at Johns Hopkins University and the University of Basel, and the appropriate consent was obtained. Specimens were kept anonymous and randomized and were approved for analysis at TGen by the Western Institutional Review Board.
NMD on cDNA microarray.
The hybridizations were done on microarrays containing 16,000 gene cDNAs17,18 printed in the Microarray Core of the National Human Genome Research Institute, National Institutes of Health. These cDNA microarrays were generated from cDNA clones obtained from the sequence-verified IMAGE clone collection from Research Genetics (Invitrogen). Of these clones, 75% represent genes with functional annotation and the rest represent expressed-sequence tags and hypothetical proteins. The gene description of these clones was based on the UniGene database (build 148) and the chromosomal base location of these genes was updated from the Genome Browser database at the University of California Santa Cruz. This update was done through the GenBank accession number or the IMAGE clone number.
As negative controls, we used 88 'housekeeping' genes (each printed eight times) whose expression does not vary substantially in several different tissues. For quality control, two slides per lot were hybridized for spot consistency, homogeneity and signal-to-noise ratios. Because of the dynamic state of the UniGene database, 12.5% of the 16,000 genes were represented by more than one clone in the arrays. These duplicates represent a unique internal control for spot consistency and hybridization reproducibility. We carried out fluorescence scanning of the slides and analysis of the resulting images, including data normalization, as described18.
We treated malignant and nonmalignant cell lines in a similar manner. For each cell line, we treated half of the subconfluent cells with 100 μg ml−1 of emetine dihydrochloride hydrate (Fluka) and used the rest as untreated controls. We then incubated both the treated and untreated cells at 37 °C for 10 h. After this incubation, we collected samples at the first time point (0 min). We added actinomycin D (Sigma-Aldrich; final concentration, 5 μg ml−1) to the remaining treated and untreated cells to stop new transcription. We collected samples at time points of 10 min, 30 min, 1 h, 2 h, 4 h and 8 h for both cell groups for most of the cell lines. Cell pellets were snap-frozen and mRNA was extracted by using a FastTrack kit (Invitrogen) in accordance with the manufacturer's instructions.
For each time point, we hybridized the untreated sample against the emetine-treated equivalent. We labeled untreated mRNA (4 μg) with Cy5-dUTP and emetine-treated mRNA (4 μg) with Cy3-dUTP (Amersham) as described18,19. Image analysis was done by DeArray software. Average intensities of the tumor samples were divided by the average intensities of the reference sample at each microarray spot after subtracting the background intensity18. Within-slide normalization was done with a ratio statistics method using housekeeping genes as described19. We filtered the data with a ratio quality method20, which computes a quality value for each ratio. The scale for the quality values ranges from 0 (poor) to 1 (good). We omitted all ratios with a quality value >0.5 from the subsequent analysis.
Real-time quantitative RT-PCR.
We used real-time quantitative RT-PCR to validate the overall changes in gene expression. We lysed cells and extracted total RNA with an RNeasy Mini kit (Qiagen). We generated cDNA in a RT-PCR reaction volume of 100 μl from 1 μg of total RNA by the ThermoScript RT-PCR cDNA synthesis method (Invitrogen). Using the gene-specific Assay-on-Demand TaqMan assay (PE/Applied Biosystems), consisting of a specific fluorogenic probe and a pair of oligonucleotides, we ran standard quantitative RT-PCR reactions for EPHB2, TP53, MLH1, ACTB and GAPD on an Opticon 2 real-time quantitative PCR instrument (MJ Research). The reactions for the quantitative RT-PCR application were done in triplicate in a 20-μl reaction volume in a 96-well plate format. The amount of total RNA in each TaqMan reaction was 10–50 ng. We normalized our data by including separate tube reactions of reference genes GAPD and ACTB. Values from emetine-treated cells at individual time points were normalized relative to untreated controls in the presence of reference genes.
CGH on cDNA microarray.
We carried out CGH on 13K Human 1 cDNA microarray slides (Agilent Technologies) as described2,3,20,21 with slight modifications. We used DNA from healthy male individuals as a reference. Genomic DNA (20 μg) was digested overnight with _Alu_I and _Rsa_I (Life Technologies). We purified digested DNAs by phenol-chloroform extraction. We labeled digested tumor DNA (6 μg) and reference DNA (6 μg) with Cy5-dUTP and Cy3-dUTP (Amersham Biosciences), respectively, in a random priming reaction using a Bioprime Labeling kit (Life Technologies). Hybridization and washes were done as described3. Microarrays were scanned by a laser confocal scanner (Agilent Technologies), and we used Feature Extraction software to measure the fluorescence intensities at the target locations (Agilent Technologies).
Data analysis.
We created a custom database (FileMaker Pro 5.0v3) including both the genomic sequence alignment information for all available mRNA sequences according to the University of California at Santa Cruz assembly (December 2001 freeze and more recent updates) and the UniGene information obtained from build 146. We entered the intensity (NMD) ratios and ratio quality values from all cDNA microarray hybridizations were into the database and then normalized the intensity ratios of each prostate cancer cell line against the average intensity ratios obtained from three nonmalignant cell lines (to produce the normalized NMD ratio) for each clone on the array.
To prioritize the candidate genes for mutational analysis, we first selected genes with a normalized NMD ratio above 3 and whose ratio quality in that cell line was above 0.5. In addition, we selected the ratios for these genes in normal control cell lines to be below 2 to filter out non–mutation-induced changes in expression. The second prioritization criterion included information about the deletion status of these genes, as measured by CGH. To plot the CGH profiles, we used a moving mean ratio of 30 consecutive clones. On the basis of normal variation in the control hybridization, mean intensity ratios below 0.9 were considered losses. Depending on the genomic region to which each gene mapped and the cumulative copy number estimate for the clones in that loci, we scored genes as being or not being in a deleted region (Fig. 2).
Mutation analysis.
We amplified DNA specimens using a standard PCR protocol and intronic primer pairs with M13 tails (sequences available on request). We purified the PCR products using a QiaQuick PCR purification kit on a BioRobot 8000 Automated Nucleic Acid Purification and Liquid Handling system (Qiagen). Quarter-volume cycle-sequencing reactions were prepared in a 96-well format using standard M13 forward or reverse primers with Big Dye Terminator Chemistry (PE/Applied Biosystems). After Sephadex purification, we separated sequence products on an ABI 3700 or ABI 3730 Capillary DNA Analyzer (PE/Applied Biosystems) in accordance with the manufacturer's protocols. Sequence chromatograms were aligned and analyzed by Sequencher version 4.1 (Gene Codes).
Colony formation assay.
We purchased the human clone of EphB2 from OriGene Technologies and subcloned it into the pIRES-dsRed2 expression vector (BD-Biosciences). We selected two subclones (A and B) and validated their full-length wild-type sequences. The mouse Ephb2 expression construct was a gift from T. Pawson (University of Toronto, Canada), and the p53 expression construct was a gift from M. Srivastava (USUHS, Bethesda, Maryland).
We transfected cells with 1.0 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's instructions except for the following modification. We treated cells (1.75 × 105) with trypsin and plated them with DNA-lipid complex in duplicate wells. After 24 h, we added 0.5 μg ml−1 of medium containing G418 to the wells and changed the medium every 2 d. After 14 d, we added Cell Titer Blue (Promega) reagent to the wells to measure cell proliferation in accordance with the manufacturer's instructions. Data were normalized to the vector control (pIRES-dsRed2) and are presented as a percentage of control proliferation. We measured colony formation after the Cell Titer Blue assay by removing medium and fixing cells with 2% paraformaldehyde for 15 min. After two washes, we stained cell colonies with Giemsa stain for 30 min. Colonies (>1.0 mm) were visually scored independently by two different individuals.
URLs.
The TP53 Cell Line Mutational Database is available at http://p53.curie.fr/. The University of California at Santa Cruz Genome Browser is available at http://genome.ucsc.edu/. Information on UniGene build 146 is available from the UniGene database at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene. Microarray data are available at the National Center for Biotechnology Information Gene Expression Omnibus website (http://www.ncbi.nlm.nih.gov/geo/).
Accession numbers.
GenBank: Homo sapiens EphB2 (EPHB2) transcript variant 1 mRNA, NM_017449; Homo sapiens EphB2 (EPHB2) transcript variant 2 mRNA, NM_004442. GEO: microarray platform, GPL1301; DU 145 time course, GSE1487; LNCaP time course, GSE1488; PC3 time course, GSE1489; GM00156 time course, GSE1490.
Note: Supplementary information is available on the Nature Genetics website.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
- Noensie, E.N. & Dietz, H.C. A strategy for disease gene identification through nonsense-mediated mRNA decay inhibition. Nat. Biotechnol. 19, 434–439 (2001).
Article CAS Google Scholar - Pollack, J.R. et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 23, 41–46 (1999).
Article CAS Google Scholar - Hyman, E. et al. Impact of DNA amplification on gene expression patterns in breast cancer. Cancer Res. 62, 6240–6245 (2002).
CAS PubMed Google Scholar - Knudson, A.G. Jr. Hereditary cancer, oncogenes, and antioncogenes. Cancer Res. 45, 1437–1443 (1985).
CAS PubMed Google Scholar - Losson, R. & Lacroute, F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl. Acad. Sci. USA 76, 5134–5137 (1979).
Article CAS Google Scholar - Culbertson, M.R. RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet. 15, 74–80 (1999).
Article CAS Google Scholar - Gibbs, M. et al. Evidence for a rare prostate cancer-susceptibility locus at chromosome 1p36. Am. J. Hum. Genet. 64, 776–787 (1999).
Article CAS Google Scholar - Kullander, K. & Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell. Biol. 3, 475–486 (2002).
Article CAS Google Scholar - Mellitzer, G., Xu, Q. & Wilkinson, D.G. Control of cell behaviour by signalling through Eph receptors and ephrins. Curr. Opin. Neurobiol. 10, 400–408 (2000).
Article CAS Google Scholar - Wang, H.U., Chen, Z.F. & Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741–753 (1998).
Article CAS Google Scholar - Gale, N.W. et al. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev. Biol. 230, 151–160 (2001).
Article CAS Google Scholar - Wilkinson, D.G. Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci. 2, 155–164 (2001).
Article CAS Google Scholar - Knoll, B. & Drescher, U. Ephrin-As as receptors in topographic projections. Trends Neurosci. 25, 145–149 (2002).
Article CAS Google Scholar - Barrios, A. et al. Eph/Ephrin signaling regulates the mesenchymal-to-epithelial transition of the paraxial mesoderm during somite morphogenesis. Curr. Biol. 13, 1571–1582 (2003).
Article CAS Google Scholar - Batlle, E. et al. β-Catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111, 251–263 (2002).
Article CAS Google Scholar - Elowe, S., Holland, S.J., Kulkarni, S. & Pawson, T. Downregulation of the Ras–mitogen-activated protein kinase pathway by the EphB2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Mol. Cell. Biol. 21, 7429–7441 (2001).
Article CAS Google Scholar - DeRisi, J. et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 14, 457–460 (1996).
Article CAS Google Scholar - Mousses, S. et al. Gene expression analysis by cDNA microarrays. in Functional Genomics (eds. Livesey, F. & Hunt, S.P.) 113–137 (Oxford University Press, Oxford, 2000).
Google Scholar - Chen, Y., Dougherty, E. & Bittner, M. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J. Biomed. Optics 2, 364–374 (1997).
Article CAS Google Scholar - Chen, Y. et al. Ratio statistics of gene expression levels and applications to microarray data analysis. Bioinformatics 18, 1207–1215 (2002).
Article CAS Google Scholar - Monni, O. et al. Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc. Natl. Acad. Sci. USA 98, 5711–5716 (2001).
Article CAS Google Scholar
Acknowledgements
We thank P. Meltzer, G. Batist, M. Kandouz, J. Khan, I. Andrulis, N. Gokgoz and E. Bruckheimer for discussions; J. Lueders, M. White and R. Autio for technical assistance; and the Sequencing Core in TGen for their services.
Author information
Authors and Affiliations
- Translational Genomics Research Institute, Cancer Drug Development Laboratory, 20 Firstfield Road, Suite 110, Gaithersburg, 20878, Maryland, USA
Pia Huusko, Jeff A Kiefer, David O Azorsa, Sukru Tuzmen, Don Weaver, Olli-P Kallioniemi & Spyro Mousses - Genetic Basis of Human Disease Research Division, Translational Genomics Research Institute, 400 North Fifth Street, Suite 1600, Phoenix, 85004, Arizona, USA
Damaris Ponciano-Jackson, Christiane Robbins, Tracy Moses, Jeffrey M Trent & John D Carpten - Medical Biotechnology, VTT Technical Research Centre of Finland, and University of Turku, PO Box 106, Turku, 20521, Finland
Maija Wolf & Olli-P Kallioniemi - Cancer Genetics Branch, National Human Genome Research Institute/NIH, 50 South Drive, Bethesda, 20892, Maryland, USA
Minna Allinen, Yidong Chen & Abdel Elkahloun - Institute of Signal Processing, Tampere University of Technology, PO Box 553, Tampere, 33101, Finland
Sampsa Hautaniemi - Department of Oncology, Jewish General Hospital, 3755 Cote St. Catherine Road, Montreal, H3T 1E2, Quebec, Canada
Mark Basik - Departments of Urology and Pathology, Johns Hopkins University School of Medicine, 600 North Wolfe Street, Baltimore, 21287, Maryland, USA
G Steven Bova - Institute of Pathology, University of Basel, Schonbeinstrasse 40, Basel, CH-4003, Switzerland
Lukas Bubendorf, Alessandro Lugli & Guido Sauter - Laboratory of Cancer Genetics, Institute of Medical Technology, University of Tampere and Tampere University Hospital, Biokatu 8, Tampere, 33520, Finland
Johanna Schleutker - Samuel Lunenfeld Research Institute, Mount Sinai Hospital, 600 University Avenue, Toronto, M5G 1X5, Ontario, Canada
Hilmi Ozcelik, Sabine Elowe & Tony Pawson
Authors
- Pia Huusko
You can also search for this author inPubMed Google Scholar - Damaris Ponciano-Jackson
You can also search for this author inPubMed Google Scholar - Maija Wolf
You can also search for this author inPubMed Google Scholar - Jeff A Kiefer
You can also search for this author inPubMed Google Scholar - David O Azorsa
You can also search for this author inPubMed Google Scholar - Sukru Tuzmen
You can also search for this author inPubMed Google Scholar - Don Weaver
You can also search for this author inPubMed Google Scholar - Christiane Robbins
You can also search for this author inPubMed Google Scholar - Tracy Moses
You can also search for this author inPubMed Google Scholar - Minna Allinen
You can also search for this author inPubMed Google Scholar - Sampsa Hautaniemi
You can also search for this author inPubMed Google Scholar - Yidong Chen
You can also search for this author inPubMed Google Scholar - Abdel Elkahloun
You can also search for this author inPubMed Google Scholar - Mark Basik
You can also search for this author inPubMed Google Scholar - G Steven Bova
You can also search for this author inPubMed Google Scholar - Lukas Bubendorf
You can also search for this author inPubMed Google Scholar - Alessandro Lugli
You can also search for this author inPubMed Google Scholar - Guido Sauter
You can also search for this author inPubMed Google Scholar - Johanna Schleutker
You can also search for this author inPubMed Google Scholar - Hilmi Ozcelik
You can also search for this author inPubMed Google Scholar - Sabine Elowe
You can also search for this author inPubMed Google Scholar - Tony Pawson
You can also search for this author inPubMed Google Scholar - Jeffrey M Trent
You can also search for this author inPubMed Google Scholar - John D Carpten
You can also search for this author inPubMed Google Scholar - Olli-P Kallioniemi
You can also search for this author inPubMed Google Scholar - Spyro Mousses
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toOlli-P Kallioniemi or Spyro Mousses.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Huusko, P., Ponciano-Jackson, D., Wolf, M. et al. Nonsense-mediated decay microarray analysis identifies mutations of EPHB2 in human prostate cancer.Nat Genet 36, 979–983 (2004). https://doi.org/10.1038/ng1408
- Received: 20 January 2004
- Accepted: 22 June 2004
- Published: 08 August 2004
- Issue Date: 01 September 2004
- DOI: https://doi.org/10.1038/ng1408