BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt–β-catenin signaling (original) (raw)
Wnt signaling through β-catenin promotes cell proliferation6,7, and abnormal activation of β-catenin due to deregulated Wnt signaling leads to adenomatous polyposis coli in intestines6,7. The BMP pathway also has a key role during gastrointestinal development8,9 and in maintaining tissue homeostasis in the adult10. According to current models, BMP2 and BMP4 function by binding to their type II receptor and recruiting type I receptors (Bmpr1a or Bmpr1b). The signal is transduced from the cytoplasm into the nucleus through Smad transcription factors10. Activity of BMP can be further regulated by its antagonist, Noggin (encoded by Nog)11. Mutations in the tumor-suppressor gene PTEN (a dual protein and lipid phosphatase)12 have also been found in gastrointestinal cancers13. PTEN is a negative regulator of phosphatidylinosital-3 kinase (PI3K)14, which, through Akt (a serine-threonine kinase), promotes cell-cycle progression and suppresses apoptosis. Thus, PTEN inhibits Akt activity and the signals downstream of Akt13.
Intestinal stem cells (ISCs), identified using modified 5-bromodeoxyuridine (BrdU)-retaining assays15, are located at the fourth or fifth cell position from the base of each crypt in the small intestine, superior to Paneth cells (Fig. 1a–c). Multipotent ISCs are responsible for generating the entire crypt-villus structure as shown by clonal assays15,16. To investigate the role of BMP signaling in regulating intestinal development, we determined the expression patterns of BMP4, its antagonist Noggin, Bmpr1a and phosphorylated (P-) Smad1, P-Smad5 and P-Smad8 (Fig. 1d–k). BMP4 was expressed in the intravillus and intercrypt mesenchymal cells, including those adjacent to ISCs (Fig. 1d,e). Bmpr1a had a gradient distribution in epithelial cells along the crypt-villus axis and was also highly expressed in ISCs (Fig. 1f,g) but not in the cells in the proliferating zone (Fig. 1f). Noggin was expressed in the submucosal region adjacent to the crypt bottom and in only some cells in the ISC position or the surrounding cells (Fig. 1h,i). The presence of active BMP signaling was reflected by the presence of P-Smad1, P-Smad5 and P-Smad8 in both villi and ISCs (Fig. 1j,k). Noggin antagonizes BMP signaling to regulate the stem cell niche during neurogenesis17. We hypothesized that transient expression of Noggin (Fig. 1h,i) may also function to control ISC properties through regulation of BMP activity.
Figure 1: Analyses of intestinal expression patterns of BMP signaling molecules.
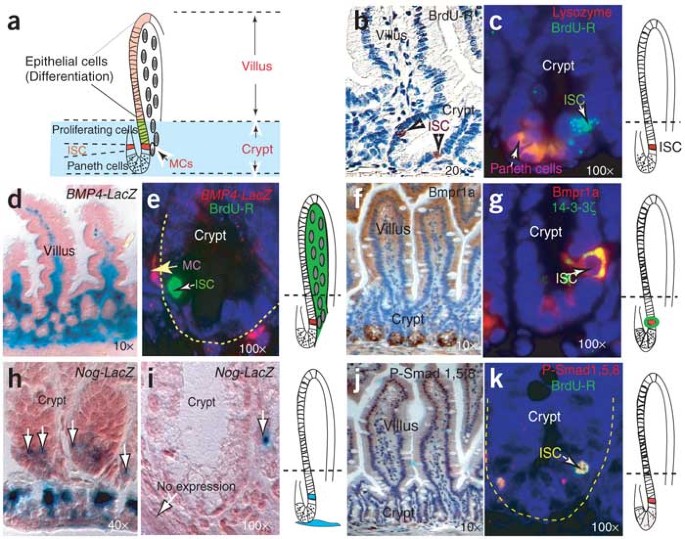
(a) Illustration of crypt-villus structure and the positions of stem, Paneth, differentiated epithelial and mesenchymal cells (MCs). (b,c) BrdU retention (BrdU-R) 22 d after treatment labels ISCs, located above the Paneth cells, which are stained by antibody to lysozyme. (d,e) BMP4 is detected (by the presence of galactosidase) in the mesenchymal cells (MC), particularly those adjacent to ISCs recognized by BrdU retention (BrdU-R). (f,g) Bmpr1a is found at varying levels in epithelial cells in the different crypt-villus regions and in the ISCs. Due to incompatibility of antibody staining, Bmpr1a was costained with 14-3-3ζ, which specifically stains cells recognized with P-PTEN (data not shown), an ISC marker (see Fig. 3f). (h) When expressed, Noggin is restricted to the basement membrane adjacent to the crypt and in some ISCs or their surrounding cells (white arrows). (i) Noggin expression is transient. (j,k) Distribution of P-Smad1, P-Smad5 and P-Smad8 along the villus-crypt axis and in ISCs. BrdU-R, BrdU retention.
Because the null mutation of Bmpr1a is lethal to the embryo18, we generated mice in which Bmpr1a can be conditionally inactivated using the interferon-inducible Mx1-cre line19. The efficiency of the Mx1-cre line in mediating _lox_P-dependent DNA excision in the intestine was determined using Z/EG reporter mice20. Expression of interferon in Mx1-cre mice was induced by injection of polyinosinic-polycytidylic acid (PolyI:C). Clonal expression of enhanced green fluorescent protein (EGFP) was detected in the entire crypt-villus of some units, consistent with previous reports15,16. As each unit is generated from a single ISC, clonal expression of EGFP indicates that the PolyI:C-induced gene deletion occurred in the stem cells. In contrast, the cells emanating from ISCs that escape targeting retain LacZ expression (Fig. 2a,b).
Figure 2: Test of the efficiency of PolyI:C-induced gene deletion and polyposis phenotype.
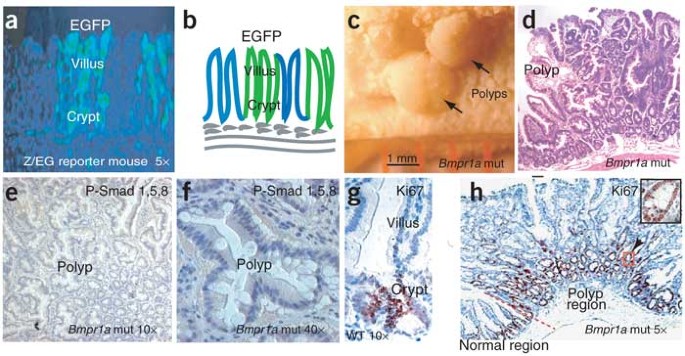
(a,b) Targeting efficiency was assayed in Mx1-cre Z/EG chimeric mice. (c,d) Polyps in small intestine and the corresponding histological sections. (e,f) Loss of expression of P-Smad1, P-Smad5 and P-Smad8. (g,h) Ki67 staining to locate the proliferation zone in intestinal sections from wild-type (WT) and Bmpr1a mutant (mut) mice.
The 15 Bmpr1a mutant mice that we analyzed all developed multiple polyps (15–40 polyps in the segment located 15–25 cm from the pylorus; Fig. 2c,d), and successful targeting of Bmpr1a was confirmed by loss of P-Smad1, P-Smad5 and P-Smad8 (Fig. 2e,f). The polyps ranged from diminutively small and sessile (averaging 0.2 mm in diameter) to large and pedunculated (averaging 2 mm in diameter). Histopathological analyses of early-stage polyps in the intestinal tract showed general overgrowth of intestinal tissue with an increased number of crypts and fused villi. At later stages, the polyps developed into structures with cystically dilated and distorted glands, some filled with mucin and inflammatory cells with surrounding fibrous stroma (Fig. 2d). These late-stage polyps had a histology similar to that seen in human juvenile polyposis syndrome1.
We observed severe gastrointestinal defects in the Bmpr1a mutant mice, which prompted us to determine how intestinal development is affected. In wild-type mice, proliferating cells were located in the upper crypt region where Bmpr1a is absent (compare Fig. 1f with Fig. 2g), as identified by Ki67 presence. In Bmpr1a mutant mice, the proliferating cell population was expanded substantially, primarily owing to the increase in the number of crypts in the polyp region (Fig. 2h). This result suggests that BMP signaling restricts crypt cell fate.
Wnt signaling normally favors crypt cell fate over villus differentiation6,7,9. The increased number of crypts suggested that Wnt signaling might be affected in the intestines of Bmpr1a mutant mice. We therefore compared Wnt signaling, as measured by the level of activated (phosphorylated) LRP6 (P-LRP6), in wild-type and Bmpr1a mutant mice. LRP6, a coreceptor for Wnt, becomes phosphorylated upon Wnt binding21 and serves as a marker for active Wnt signaling. Wnt signaling was active in normal intestinal crypts (Fig. 3a) and in the polyp crypts of Bmpr1a mutant mice (Fig. 3b). We also measured the transcriptional activity of β-catenin by electroporating, into cultured intestinal segments, a reporter plasmid bearing TCF–β-catenin binding sites (Top)22 driving a short-lived mutant of green fluorescent protein (GFP; _Top_-d2GFP). β-catenin transcriptional activity was detected primarily in the ISCs, despite the widespread presence of an active Wnt signal (as measured by P-LRP6) in the progenitor cells of crypts (Fig. 3c,d). This observation led us to hypothesize that Wnt signaling is required for the nuclear activity of β-catenin but is not sufficient to fully activate β-catenin in ISCs, particularly when BMP signaling is active (Fig. 1k).
Figure 3: Expression of P-LRP6, P-PTEN, P-Akt and β-catenin in crypts and stem cell compartment in intestinal segments, and detection of transcriptional activity of β-catenin.
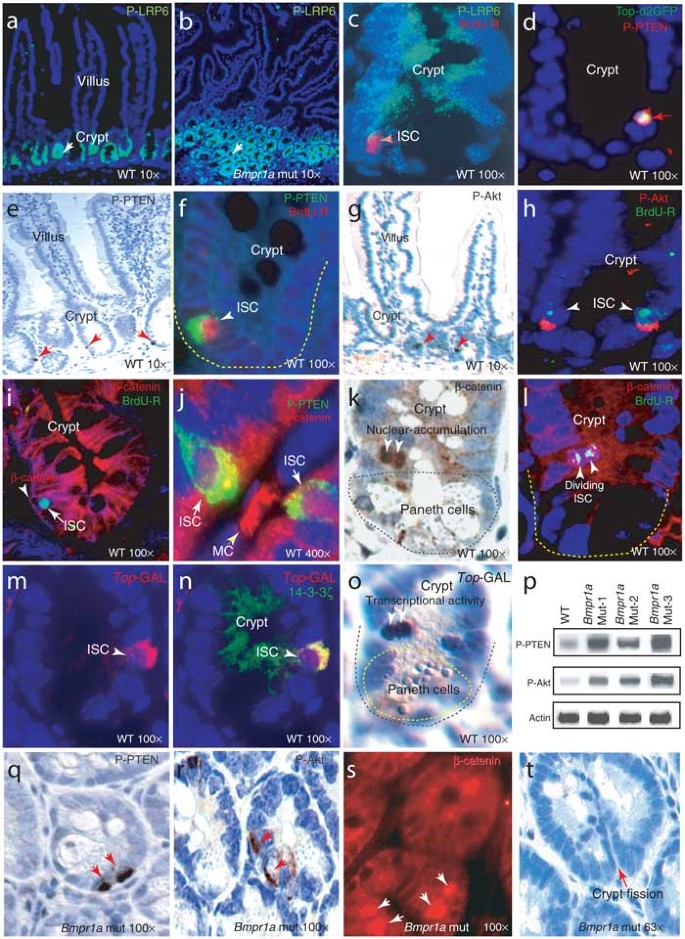
(a,b) LRP6 is predominantly detected in crypts of wild-type and mutant intestines (white arrow). (c) Costaining of P-LRP6 with BrdU retention in ISCs. (d) Detection of _Top_-d2GFP in P-PTEN+ ISCs. (e–h) Detection of P-PTEN and P-Akt in ISCs and costaining of cells retaining BrdU (BrdU-R) with P-PTEN or P-Akt in small intestine. (i,j) Costaining of β-catenin with BrdU retention (BrdU-R) and P-PTEN. (k,l) Detection of nuclearly accumulated β-catenin in dividing ISCs, recognized by BrdU retention (BrdU-R). (m–o) Detection of transcriptional activity of β-catenin in ISCs in _Top_-GAL transgenic mice. Antibody to galactosidase was used to stain a frozen section. Due to incompatibility of antibodies, ISCs were recognized by 14-3-3ζ, which specifically costains P-PTEN. (p) Western-blot assays to measure the protein levels of P-PTEN and P-Akt in intestines of wild-type and Bmpr1a mutant mice. (q–s) Detection of stem cell duplication in polyp region of intestines of Bmpr1a mutant mice, as recognized by P-PTEN, P-Akt and nuclearly-localized β-catenin, respectively. (t) Representation of crypt fission in polyp region of intestines of Bmpr1a mutant mice. mut, Bmpr1a mutant mice; WT, wild-type mice.
Inactivation of PTEN has been reported to activate Akt and the subsequent nuclear accumulation of β-catenin23, suggesting that the PTEN-Akt pathway might regulate β-catenin activity in ISCs. We therefore analyzed both PTEN and the inactive (phosphorylated) form of PTEN (P-PTEN)24 in intestines of wild-type mice and found that P-PTEN was present specifically in ISCs (Fig. 3e). We detected P-PTEN in BrdU-retaining ISCs (Fig. 3f), supporting this conclusion. We also detected phosphorylated Akt (P-Akt) in BrdU-retaining ISCs (Fig. 3g,h), consistent with the role of PTEN as a negative regulator of Akt14. We then asked whether the PTEN-Akt pathway affects β-catenin activity in ISCs. We observed that β-catenin was associated with the membrane in quiescent ISCs (Fig. 3i) but was found in the nucleus of P-PTEN+ ISCs (Fig. 3j). This nuclear localization of β-catenin correlates with the activation of _Top_-d2GFP reporter plasmid in P-PTEN+ ISCs (Fig. 3d). We also found nuclearly localized β-catenin in dividing (BrdU-retaining) ISCs (Fig. 3k,l). Furthermore, analysis of _Top_-GAL25 transgenic mice showed that in vivo β-catenin signaling was active in ISCs, including those actively dividing (Fig. 3m–o). Taken together, these data support the idea that nuclearly localized β-catenin has a role in promoting ISC division. Thus, inactive PTEN (P-PTEN), through Akt, may coordinate with the Wnt signal to fully activate β-catenin in ISCs.
We next asked what signal controls PTEN activity. Genetic evidence suggests that there is an association between the BMP and PTEN pathways4, and the BMP signal has been shown to enhance PTEN activity5. Western-blot analysis showed that levels of P-PTEN and P-Akt were 4–5 times higher in three Bmpr1a mutant mice than in wild-type mice (Fig. 3p), suggesting that BMP inhibits Akt activity through regulation of PTEN activity. We further examined the levels of P-PTEN and P-Akt in ISCs when BMP signaling is blocked. We detected only a single ISC in the ISC position in wild-type crypts (Fig. 3e–h) but detected two side-by-side, aligned ISCs in a portion of the crypts in the polyp regions in Bmpr1a mutant mice, as identified by the presence of P-PTEN or P-Akt (Fig. 3q,r; see below for ISC number quantification). This observation indicates that BMP signaling regulates the PTEN-Akt pathway in ISCs. We considered whether β-catenin activity changed correspondingly in Bmpr1a mutant mice. The presence of doublet cells showing nuclear localization of β-catenin (Fig. 3s) in the polyp region further supports the interpretation that nuclear accumulation of β-catenin in ISCs is inhibited by BMP signaling by regulating PTEN activity. We also observed crypt fission in the polyp region of Bmpr1a mutant mice (Fig. 3t).
Therefore, a second signal must abrogate BMP and coordinate with Wnt to fully activate β-catenin in ISCs. To address this, we also analyzed the effect of Noggin on levels of P-PTEN and P-Akt and on the subcellular localization of β-catenin in ex vivo cultured intestine (Fig. 4a–l). Treatment with Noggin increased the levels of P-PTEN and P-Akt and led to the relocation of β-catenin to the nucleus (Fig. 4c,g,k), although western blotting showed that the total amount of β-catenin in cells did not change substantially (Supplementary Fig. 1 online). A PI3K inhibitor, Ly294002, antagonized the effect of Noggin on nuclear relocalization of β-catenin (Fig. 4l), arguing that this regulation is mediated through the PTEN-Akt pathway.
Figure 4: Analysis of PTEN, Akt and β-catenin activity.
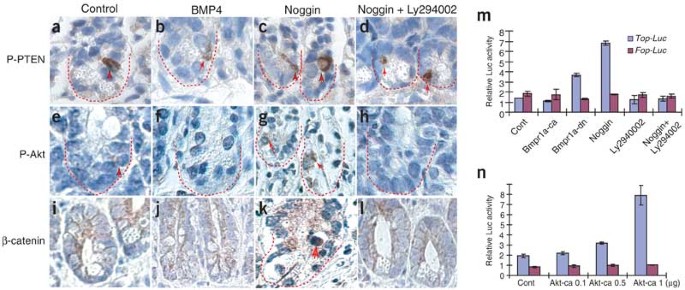
(a–l) Analysis of changes in expression of P-PTEN and P-Akt, and cellular localization of β-catenin in the crypt under different conditions in ex vivo cultured intestinal segments. Red arrows indicate the detected molecules in the stem cell compartment. (m,n) Transfection assay to test the regulation of β-catenin activity by constitutively activated Bmpr1a (Bmpr1a-ca), dominant-negative Bmpr1a (Bmpr1a-dn), Noggin and constitutively active Akt (Akt-ca) using Top-Luc reporters.
Further supporting the idea that BMP has a role in regulating β-catenin transcriptional activity through PTEN and Akt, exposure to Noggin or expression of dominant-negative Bmpr1a activated β-catenin activity, whereas expression of constitutively activated Bmpr1a or treatment with Ly294002 inhibited β-catenin activity as assayed by Top-Luc (luciferase) activity (Fig. 4m). We also tested the direct regulation of β-catenin activity by Akt. Constitutively active Akt also enhanced β-catenin activity in a dosage-dependent manner (Fig. 4n). These results provide further evidence supporting our hypothesis that β-catenin activity can be regulated by BMP signaling through Bmpr1a and the PI3K-Akt pathway.
BMP signaling may therefore have a role in inhibiting ISC self-renewal by inhibiting β-catenin activity, thereby balancing the role of Wnt–β-catenin in promoting stem cell self-renewal26. If this hypothesis is correct, then we would anticipate an increase in the self-renewal capacity of ISCs when the BMP signal is blocked. We therefore examined the stem cell zone in multiple crypts of the polyp regions in Bmpr1a mutant mice. We compared the number of ISCs in equal lengths (670 μm) of intestinal sections from nine normal regions of three wild-type mice and five polyp regions of three Bmpr1a mutant mice (Table 1); ISCs were recognized by P-PTEN. We found that, on average, there were ∼3.1 times more crypts in polyp regions (47 ± 3.8 crypts; mean ± s.d.) than in normal regions (14.8 ± 1.1 crypts; mean ± s.d.) of intestines of Bmpr1a mutant mice. This increase in crypt number was accompanied by a ∼1.6-fold increase in the number of ISCs per crypt. This resulted in a ∼5-fold increase in the average number of ISCs in Bmpr1a mutant mice compared with wild-type mice. Taken together, these results suggest that BMP signaling has a role in inhibiting ISC self-renewal by regulating PTEN activity, consistent with previous reports that inactivation of PTEN leads to activation of PI3K-Akt and results in expansion of embryonic and neural stem cell populations27,28.
Table 1 Quantification of ISCs
BMP signaling was recently reported to repress crypt formation and polyp growth in Nog transgenic mice by inhibiting Wnt signaling9. Using a Bmpr1a mutant model, we obtained similar findings and further explored the underlying mechanisms. We are not excluding the possibility that inactivation of Bmpr1a affects the canonical BMP-Smad pathway, which may control expression of Wnt or Frizzled proteins and may also regulate epithelial differentiation and apoptosis. That pathway may, in part, contribute to the development of polyposis (as mutations in SMAD4 also lead to polyp formation)2. But our data clearly show that PTEN, through PI3K-Akt, mediates the convergence of the BMP and Wnt pathways on control of β-catenin activity, thereby ensuring a balanced control of ISC self-renewal (Supplementary Fig. 2 online). Wnt signaling, as detected by P-LRP6, is active not only in stem cells but in all the proliferating cells. We also found that nuclearly localized β-catenin and the underlying transcriptional activity of β-catenin are primarily associated with P-PTEN in ISCs. These observations suggest that Wnt signaling is required for stem cell activation and self-renewal but is not sufficient. A second signal is therefore required for stem cell activation. We propose that this second signal is dependent on the transient expression of Noggin and is required to override the BMP signal, releasing the inhibition of β-catenin by PTEN and thus coordinating with Wnt signaling to activate stem cells (Supplementary Fig. 2 online). Supporting this idea, the Noggin-Wnt coordination has also been reported to be required for initiation of the hair growth cycle29. Finally, it has been proposed that crypt fission (duplication) is triggered by stem cell duplication30. In this study, we document the correlation between stem cell duplication (Fig. 3q–s) and crypt number increase, suggesting that BMP signaling may control crypt fission (Fig. 3t) through inhibition of stem cell self-renewal and their subsequent duplication.
Note: Supplementary information is available on the Nature Genetics website.