Genetic variation, classification and 'race' (original) (raw)
Few concepts have as tarnished and contentious a history as 'race'1. Among both the scientific and lay communities, the notion that humans can be grouped into different races has been enshrined by some and dismissed by others. Even the definition of race varies considerably, depending on context and criteria2,3. Nevertheless, race continues to be used in a variety of applications. Forensic databases in the US are typically organized according to traditional racial and ethnic categories (e.g., African-American, European-American, Hispanic). Investigators funded by the US National Institutes of Health are required to show that minority populations are adequately represented in biomedical studies. Responses to medical therapies, such as drugs, are often compared among populations that are divided according to traditional racial divisions. Among the general public, the validity of racial categories is often taken for granted.
Not surprisingly, biomedical scientists are divided in their opinions about race. Some characterize it as “biologically meaningless”4 or “not based on scientific evidence”5, whereas others advocate the use of race in making decisions about medical treatment or the design of research studies6,7,8. Amid this controversy, modern human genetics has generated a staggering array of new data. For the first time, it is possible to study human genetic variation using not just a few dozen polymorphisms, but hundreds or even thousands. In addition to neutral polymorphisms that inform us about population history, increasing numbers of variants that contribute to disease are being discovered.
In this review, we provide a general overview of patterns of human variation, first at the population level and then at the individual level. We also discuss whether current genetic data support traditional concepts of race, and the implications of these data for medical research and practice.
Variation at the population level
The average proportion of nucleotide differences between a randomly chosen pair of humans (i.e., average nucleotide diversity, or π) is consistently estimated to lie between 1 in 1,000 and 1 in 1,500 (refs. 9,10). This proportion is low compared with those of many other species, from fruit flies to chimpanzees11,12, reflecting the recent origin of our species from a small founding population13. The π value for Homo sapiens can be put into perspective by considering that humans differ from chimpanzees at only 1 in 100 nucleotides, on average14,15. Because there are approximately three billion nucleotide base pairs in the haploid human genome, each pair of humans differs, on average, by two to three million base pairs.
Of the 0.1% of DNA that varies among individuals, what proportion varies among main populations? Consider an apportionment of Old World populations into three continents (Africa, Asia and Europe), a grouping that corresponds to a common view of three of the 'major races'16,17. Approximately 85–90% of genetic variation is found within these continental groups, and only an additional 10–15% of variation is found between them18,19,20 (Table 1). In other words, ∼90% of total genetic variation would be found in a collection of individuals from a single continent, and only ∼10% more variation would be found if the collection consisted of Europeans, Asians and Africans. The proportion of total genetic variation ascribed to differences between continental populations, called FST, is consistent, regardless of the type of autosomal loci examined (Table 1). FST varies, however, depending on how the human population is divided. If four Old World populations (European, African, East Asian and Indian subcontinent) are examined instead of three, FST (estimated for 100 Alu element insertion polymorphisms) decreases from 14% to 10% (ref. 21). These estimates of FST and π tell us that humans vary only slightly at the DNA level and that only a small proportion of this variation separates continental populations.
Table 1 Distribution of genetic variation in Old World continental populations
Allele frequencies in populations can be compared to assess the extent to which populations differ from one another. The overall pattern of these genetic distances, summarized in a network diagram (Fig. 1), shows several important trends. First, populations tend to cluster according to their geographic distance from one another. This is to be expected, as geographically distant populations were less likely to exchange migrants throughout human evolutionary history. Second, the African populations are more diverse, a pattern consistent with many studies that have compared π in various human populations22,23,24. Third, the largest genetic distance is seen between African and non-African populations. Finally, because this network is based on Alu insertion polymorphisms, it is possible to designate unambiguously the allelic state of the ancestral human population as one in which none of the insertions would be present. This allows us to 'root' the tree and shows that the root falls closest to African populations. All of these findings, which are in accord with many other studies based on different types of genetic variation assessed in different samples of humans25,26,27,28, support an evolutionary scenario in which anatomically modern humans evolved first in Africa, accumulating genetic diversity. A small subset of the African population then left the continent, probably experienced a population bottleneck and founded anatomically modern human populations in the rest of the world29. Of special importance to discussions of race, our species has a recent, common origin.
Figure 1: A neighbor-joining network of population similarities, based on the frequencies of 100 Alu insertion polymorphisms.
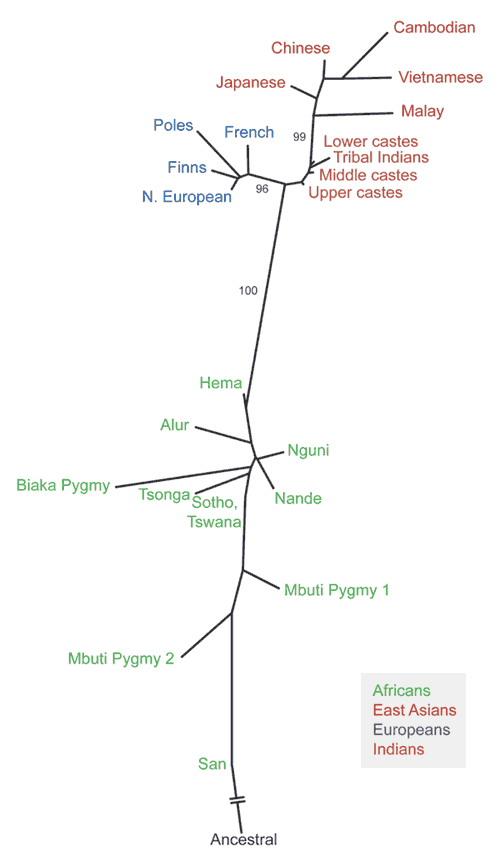
The network is rooted using a hypothetical ancestral group that lacks the Alu insertions at each locus. Bootstrap values are shown (as percentages) for main internal branches. (Because of the relatively small sample sizes of some individual populations, bootstrap values for terminal branches within main groups are usually smaller than those of the main branches, indicating less statistical support for terminal branches.) The population groups and their sample sizes are as follows: Africans (152): Alur, 12; Biaka Pygmy, 5; Hema, 18; Coriell Mbuti Pygmy, 5; a second sample of Mbuti Pygmy from the Democratic Republic of the Congo, 33; Nande, 17; Nguni, 14; Sotho/Tswana, 22; Kung (San), 15; Tsonga, 14. East Asians (61): Cambodian, 12; Chinese, 17; Japanese, 17; Malay, 6; Vietnamese, 9. Europeans (118): northern Europeans, 68; French, 20; Poles, 10; Finns, 20. South Indians (365): upper caste Brahmin, Kshatriya and Vysya, 81; middle caste Kapu and Yadava, 111; lower caste Relli, Mala and Madiga, 74; tribal Irula, Khonda Dora, Maria Gond and Santal, 99. Further details on samples and methods are given in ref. 21.
Variation at the individual level
Comparisons of populations are sometimes criticized because the allocation of individuals into groups imposes a pre-existing structure and may influence the outcome of a genetic study. Furthermore, populations are defined in many (often arbitrary) ways. Some of these objections can be overcome by comparing individuals rather than populations.
A common approach in studying individual genetic variation is to compute the genetic similarity between all possible pairs of individuals in a sample and then to search for clusters of individuals who are most similar to one another. At least two such studies have concluded that the observed clustering patterns do not correspond well to the subjects' geographic origins or 'ethnic labels'30,31. These studies, however, were based on only several dozen or fewer loci, and a small sample of unselected loci does not typically provide sufficient power to detect population structure when individuals are analyzed6,32. In contrast, studies based on more loci32,33,34 show that individuals tend to cluster according to their ancestry or geographic origin. Figure 2 shows a tree in which genetic similarity, based on 190 loci, is portrayed among individual members of most of the populations illustrated in Figure 1. The longest branches in this tree separate individuals within the same continental populations, as expected from the FST results discussed above (i.e., most variation occurs within populations). The longest internal branch separates African from non-African individuals, again in agreement with previous results at the population level. The next cluster consists entirely of Europeans, and a final cluster contains all of the East Asian subjects and one European. The robustness and validity of these findings are supported by other studies, which, despite using different loci and different population samples, obtained similar patterns34,35,36,37.
Figure 2: A neighbor-joining tree of individual similarities, based on 60 STR polymorphisms72, 100 Alu insertion polymorphisms21 and 30 restriction site polymorphisms73.
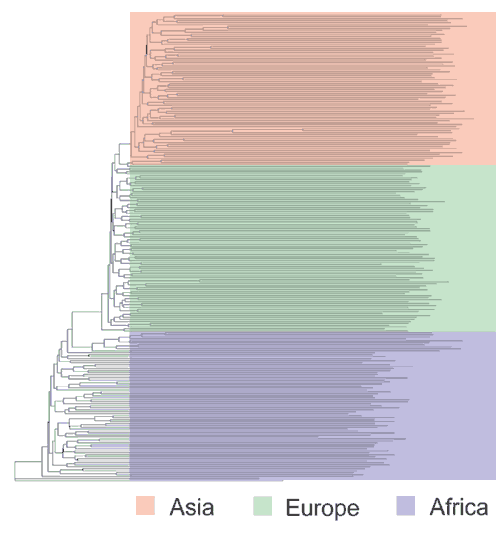
The percentage of shared alleles was calculated for all possible pairs of individuals, and a neighbor-joining tree was formulated using the PHYLIP software package74. African individuals are shown in blue, European individuals in green and Asian individuals in orange.
A statistically more sophisticated approach for cluster definition is afforded by the structure program38, in which individuals are first randomly assigned into one of k groups, without regard to population affiliation. Individuals are then moved between groups to minimize Hardy-Weinberg disequilibrium and interlocus linkage disequilibrium within each group (minimal within-groups disequilibrium ensures genetic homogeneity within each group). Posterior probabilities of membership in each of the k groups are then estimated for each individual. Figure 3a shows the results of applying the structure program to individuals from the populations shown in Figure 2. When 100 Alu insertion polymorphisms and 60 short tandem repeat (STR) polymorphisms were used, all Europeans, East Asians and Africans were correctly placed according to their respective continents of origin32.
Figure 3: Structure analysis.
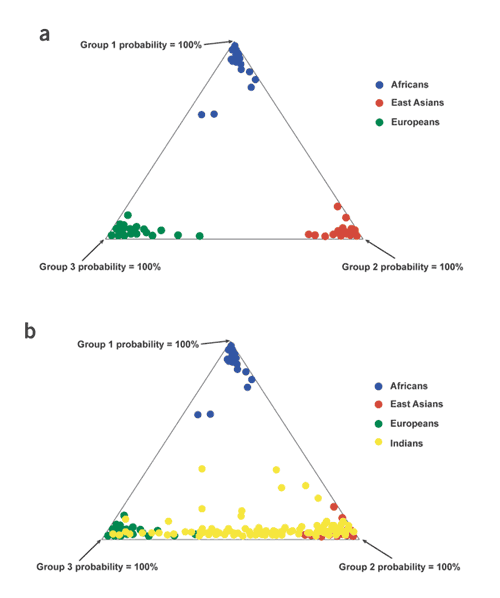
(a) Results of applying the structure program to 100 Alu insertion polymorphisms typed in 107 sub-Saharan Africans, 67 East Asians and 81 Europeans. Individuals are shown as dots in the diagram. Three clusters appear in this diagram; a cluster membership posterior probability of 100% would place an individual at an extreme corner of the diagram. (b) A second application of the structure program, using the individuals shown in a as well as 263 members of caste populations from South India. Adapted from ref. 32.
These results may seem paradoxical in light of the small proportion of genetic variation that separates continental populations (FST), but FST should not be confused with the accuracy of classification. Incorporation of additional loci in a population study has minimal effects on FST but steadily increases the accuracy of classification39. This is because FST treats each locus in isolation from the others; it takes no account of correlations among loci. The more loci there are in the data set, the more of these correlations there are, and the more information is ignored by FST.
Considering the results shown in Figures 2 and 3a, it might be tempting to conclude that genetic data verify traditional concepts about races. But the individuals used in these analyses originated in three geographically discontinuous regions: Europe, sub-Saharan Africa and East Asia. When a sample of South Indians, who occupy an intermediate geographic position (see also Fig. 1) is added to the analysis (Fig. 3b), considerable overlap is seen among these individuals and both the East Asian and European samples, probably as a result of numerous migrations from various parts of Eurasia into India during the past 10,000 years40. Thus, the South Indian individuals do not fall neatly into one of the categories usually conceived as a 'race'. In addition, examination of the posterior probabilities estimated by structure shows that most individuals in Figure 3 are not classified with 100% probability into one of the main clusters32 (similar results were obtained in ref. 33). In other words, each individual within a cluster shares most, but not all, of his or her ancestry with other members of the cluster (e.g., a member of the European cluster might have a posterior probability of 90% for assignment to that cluster, with 5% probability of assignment into each of the other two clusters in Fig. 3a). Ancestry, then, is a more subtle and complex description of an individual's genetic makeup than is race41. This is in part a consequence of the continual mixing and migration of human populations throughout history. Because of this complex and interwoven history, many loci must be examined to derive even an approximate portrayal of individual ancestry.
The picture that begins to emerge from this and other analyses of human genetic variation is that variation tends to be geographically structured, such that most individuals from the same geographic region will be more similar to one another than to individuals from a distant region. Because of a history of extensive migration and gene flow, however, human genetic variation tends to be distributed in a continuous fashion and seldom has marked geographic discontinuities19,42. Thus, populations are never 'pure' in a genetic sense, and definite boundaries between individuals or populations (e.g., 'races') will be necessarily somewhat inaccurate and arbitrary.
Though now supported by many genetic data, this concept is hardly new. Blumenbach43, writing in the 1700s, acknowledged extensive morphological overlap among populations or races. Charles Darwin, some 100 years later, wrote, “It may be doubted whether any character can be named which is distinctive of a race and is constant.”44 Scientists recognized shared genetic variation among populations more than 50 years ago, although their conclusions were based on relatively small numbers of informative loci45. This pattern of shared variation has important implications for our understanding of population differences and similarities, and it also bears on critical biomedical issues.
Genetic variation, race and medicine
Race and ethnicity have long been incorporated into medical decision-making processes. For example, physicians are typically aware that sickle-cell disease is much more common in African and Mediterranean populations than in northern European populations, whereas the reverse is true for cystic fibrosis and hemochromatosis. Although such distinctions are often clearest for single-gene diseases, perceived population differences influence the diagnosis and treatment of common diseases as well. There is evidence, for example, for population differences in response rates to drugs used in treating hypertension46 and depression47,48.
Broad population categories can be discerned genetically when enough polymorphisms are analyzed, as seen in Figure 3, so these categories are not devoid of biological meaning. When several thousand or more polymorphisms are examined, individual populations, such as Japanese and Chinese, can be delineated34, and members of 'admixed' American populations, such as Hispanics, African-Americans and European-Americans, can be accurately identified34,49. Similar results are obtained whether coding or noncoding polymorphisms are used49.
At face value, such results could be interpreted as supporting the use of race in evaluating medical treatment options. But race and ancestry are not equivalent. Many polymorphisms are required to estimate an individual's ancestry, whereas the number of genes involved in mediating a specific drug response may be relatively small50. If disease-associated alleles are common (and thus of clinical significance), they are likely to be relatively ancient and therefore shared among multiple populations51,52. Consequently, an individual's population affiliation would often be a faulty indicator of the presence or absence of an allele related to diagnosis or drug response.
To illustrate these points, consider the gene angiotensinogen (AGT), which encodes a key component of the renin-angiotensin blood-pressure regulation pathway. An AGT variant, 235T, is associated (in many populations) with a 10–20% increase in the risk of developing hypertension53. This variant has a frequency as high as 90% in some African populations and as low as 30% in European populations54. There is substantial population variation in the frequency of this allele, but it is present with an appreciable frequency in all populations examined to date. If a tree is made using allelic variation in this 14.4-kb gene (Fig. 4), the results differ markedly from those shown in Figure 2. In many cases, individuals from different continents are more similar to one another, with regard to this gene, than are individuals from the same continent.
Figure 4: A neighbor-joining tree formulated using the same methods as in Figure 2, based on polymorphisms in the 14.4-kb gene AGT.
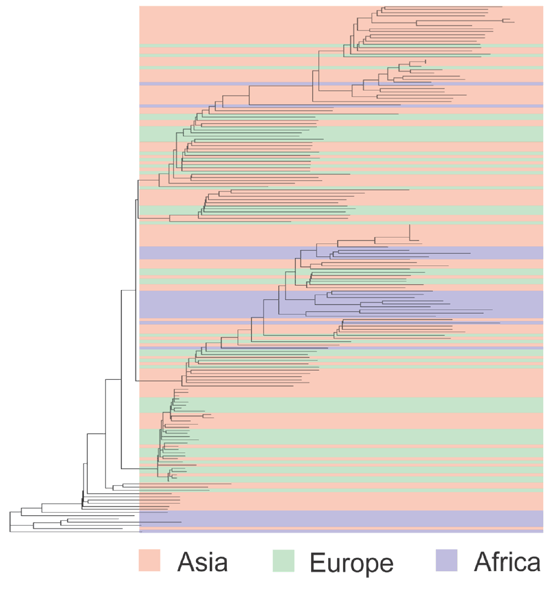
A total of 246 sequence variants, including 100 singletons, were observed. The 368 European, Asian and African individuals are described further in ref. 54.
Another example is given by the gene CYP2D6, which encodes a member of the cytochrome P450 family involved in the metabolism of many important drugs55. Null alleles of CYP2D6 render the gene product inactive. Individuals who are homozygous with respect to CYP2D6 null alleles cannot, for example, effectively convert codeine to its active form, morphine, and experience little or no analgesic effect. The median frequencies of null CYP2D6 alleles vary from 6% in Asian populations to 7% in African populations and 26% in European populations56. Thus, there is substantial population variation, but these variants are present in all populations. Population affiliation alone would be, at best, a crude and potentially inaccurate indicator of response to codeine and other CYP2D6-metabolized drugs.
Much discussion has been devoted to recent findings that hypertensive African-Americans show less response, on average, than hypertensive European-Americans to angiotensin-converting enzyme (ACE) inhibitors46,57. A recent meta-analysis showed that the average difference in systolic blood pressure reduction in African-Americans versus European-Americans was only 4.6 mm Hg, and the standard deviations of the change in blood pressure in European-Americans and African-Americans were 12 and 14 mm Hg, respectively58. Clearly, many African-Americans would respond better to ACE inhibitors than would many European-Americans. To conclude, on the basis of population averages, that ACE inhibitors are ineffective in African-Americans could deny many people a powerful and appropriate drug treatment.
Patterns such as these are seen in many genes that are thought to underlie susceptibility to common diseases. Allelic variation tends to be shared widely among populations, so race will often be an inaccurate predictor of response to drugs or other medical treatments. It would be far preferable to test directly the responsible alleles in affected individuals50,59. Such an assessment of allelic variation is a hope for the future, because most of the genes that contribute to common diseases remain to be identified. In addition, nongenetic factors nearly always have an important (and sometimes predominant60) role in disease susceptibility. Genetic assessment alone will never be a panacea. Nevertheless, progress is being made in identifying alleles that underlie complex disease susceptibility61,62,63,64,65, and rapid developments in new technologies such as microarrays promise to provide efficient and far-ranging genetic assays66.
Conclusions
Data from many sources have shown that humans are genetically homogeneous and that genetic variation tends to be shared widely among populations. Genetic variation is geographically structured, as expected from the partial isolation of human populations during much of their history. Because traditional concepts of race are in turn correlated with geography, it is inaccurate to state that race is “biologically meaningless.” On the other hand, because they have been only partially isolated, human populations are seldom demarcated by precise genetic boundaries. Substantial overlap can therefore occur between populations, invalidating the concept that populations (or races) are discrete types.
When large numbers of loci are evaluated, it is often possible to infer individual ancestry, at least approximately. If done accurately and with appropriate reservations, ancestral inference may be useful in genealogical studies, in the forensic arena and in the design of case-control studies. This should not be confused, however, with the use of ethnicity or race (genetically measured or self-identified) to make decisions about drug treatment or other medical therapies. Responses to these therapies will often involve nongenetic factors and multiple alleles, and different populations will often share these alleles. When it finally becomes feasible and available, individual genetic assessment of relevant genes will probably prove more useful than race in medical decision making.
In the meantime, ethnicity or race may in some cases provide useful information in biomedical contexts, just as other categories, such as gender or age, do. But the potential usefulness of race must be balanced against potential hazards. Ignorance of the shared nature of population variation can lead to diagnostic errors (e.g., the failure to diagnose sickle-cell disease in a European individual or cystic fibrosis in an Asian individual) or to inappropriate treatment or drug prescription. The general public, including policy-makers, are easily seduced by typological thinking, and so they must be made aware of the genetic data that help to prove it wrong.
A particular area of concern is in the genetics of human behavior. As genes that may influence behavior are identified, allele frequencies are often compared in populations67,68. These comparisons can produce useful evolutionary insights but can also lead to simplistic interpretations that may reinforce unfounded stereotypes69. In assessing the role of genes in population differences in behavior (real or imagined), several simple facts must be brought to the fore. Human behavior is complicated, and it is strongly influenced by nongenetic factors70. Thousands of pleiotropic genes are thought to influence behavior, and their products interact in complex and unpredictable ways. Considering this extraordinary complexity, the idea that variation in the frequency of a single allele could explain substantial population differences in behavior would be amusing if it were not so dangerous.
Race remains an inflammatory issue, both socially and scientifically. Fortunately, modern human genetics can deliver the salutary message that human populations share most of their genetic variation and that there is no scientific support for the concept that human populations are discrete, nonoverlapping entities. Furthermore, by offering the means to assess disease-related variation at the individual level, new genetic technologies may eventually render race largely irrelevant in the clinical setting. Thus, genetics can and should be an important tool in helping to both illuminate and defuse the race issue.