Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung (original) (raw)
Main
Differences in the severity and ultimate outcome of viral infections can relate to the dose of the virus, its route of infection and the genetics, physiological state and immune status of the host. Until recently, most discussions of the immune status centered on whether or not the host had been vaccinated against or infected with the same, or a closely related, pathogen. Such exposure can give rise to neutralizing antibodies, which prevent infection with the homologous virus from being initiated, and to memory T cells, which prevent the infection from progressing. The efficacy of memory T cells after an infection may be due to the fact that they persist at relatively high frequencies1,2,3,4,5 as slowly cycling cells6,7 in lymphoid and peripheral organs. Also, they are constitutively at a higher state of activation than naïve T cells. For example, they can be induced into cell cycle by cytokines such as interferon-α (IFN-α) or IFN-β and interleukin 15 (IL-15)8; they can also be triggered by relatively low-affinity interactions through their T cell receptors (TCRs)7,9. Even some apparently resting memory T cells can be cytolytically active5,10.
Until recently studies tended to ignore the fact that in the natural environment each individual experiences many sequential infections that give rise to memory T cell pools of significant size and complexity. It has become clear that some memory T cells can be reactivated by heterologous viral infections11,12 and that T cell cross-reactivity may be a relatively common event12,13,14,15,16. Analysis of experimental intraperitoneal (i.p.) infections in mice has suggested that memory T cell populations generated in response to one virus can indeed alter the course of disease in response to unrelated viruses17. Based on these observations we would predict that memory T cells exist in a dynamic network, the repertoire of which remains static in the absence of infection. However, each new infection will activate some of these memory T cells, modulate the original repertoire and alter the course of disease. Here, we developed these hypotheses to increase our understanding of this new and still poorly understood area of immunology, which is termed heterologous immunity.
Because the majority of viruses enter the host through the mucosa of the respiratory or gastrointestinal tract, which have highly integrated mucosal immune systems18,19, we focused our studies on a more natural respiratory mucosal model of virus infection. Developments in the identification of antigen-specific T cell responses1,2,20,21,22,23 enabled us to examine the extent of heterologous memory T cell participation in a peripheral organ, the lung, which is the initial site of virus replication, as well as the conventional lymphoid compartments. To address heterologous immunity in a setting where infection occurs via the mucosa, we infected mice intranasally with two unrelated viruses, lymphocytic choriomeningitis virus (LCMV) and vaccinia virus (VV). We first immunized mice with LCMV, an RNA virus, which is a relatively common human pathogen: 4–15% of the general population are serologically positive for LCMV and, in the natural environment, it is predominantly transmitted via the respiratory route24,25,26. In C57BL/6 mice it induces a highly defined high-frequency T cell response to several epitopes27,28,29. These include four H-2Db–restricted epitopes: LCMV nucleoprotein eptiope 396 (NP396), LCMV glycoprotein epitope 33 (GP33), GP276 and GP92 and two H-2Kb–restricted epitopes: NP205 and GP34. By immunizing mice first with LCMV, which has so many well defined epitopes, it allowed us to examine the relative ability of a heterologous virus to reactivate discrete memory T cell populations. The challenge virus was an unrelated DNA virus, VV, which can reactivate LCMV-specific cytolytic T lymphocyte (CTL) activity after i.p. infection12. VV, used for vaccination against smallpox, is a model for poxviruses, which are naturally transmitted via the respiratory mucosa30. VV has been used in a murine pneumonia model31, and human smallpox infection frequently induces pneumonitis30.
Using this respiratory model system we show here that an acute heterologous respiratory virus infection did in fact functionally activate memory CD8+ T cells specific to another virus in vivo. These T cells selectively expanded, resulting in modulation of the original T cell repertoire and modification of the lung pathology and disease outcome.
Results
Immunity to LCMV protects against VV
Respiratory LCMV infection of C57BL/6 mice via the intranasal (i.n.) route induced a potent acute CD8+ T cell response in the lungs, draining mediastinal lymph nodes (MLNs) and spleens. This led to clearance of LCMV, which was comparable to clearance of systemic i.p. LCMV infection32 (day 3: 5.3±0.08 log plaque-forming units (PFU)/lung, 5.0 log PFU/4 pooled MLNs, 5.5±0.3 log PFU/spleen; day 7: 3.6±0.08 log PFU/lung, 1.5 log PFU/4 pooled MLNs, 2.4±0.2 log PFU/spleen; day 16: no LCMV was detected in these organs; _n_=4 mice per group). To determine whether previous immunity to LCMV could provide protective heterologous immunity to VV, we examined its influence on mortality and VV titers. When mice were infected with a lethal dose of VV, there was a marked difference between the groups: 83% of the LCMV-immune mice survived compared to only 17% of the control mice (Fig. 1). Three similar mortality experiments showed that the difference in mortality between the groups was statistically significant: 94±6% of the LCMV-immune mice survived the lethal VV challenge compared to only 42±12% of the control mice (P<0.02, _n_=3). These experiments were carried out 6–18 weeks after LCMV infection, so this protection appeared to be independent of the time of VV challenge after LCMV infection.
Figure 1: Immunity to LCMV provides protection against respiratory infections with VV.
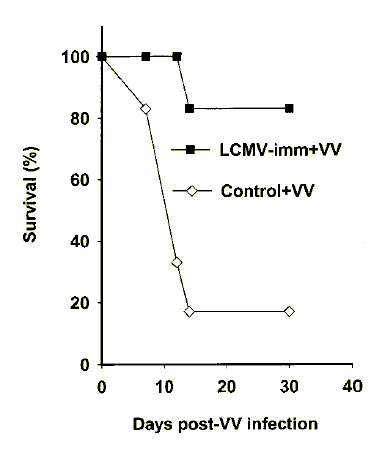
Decreased mortality of LCMV-immune mice (LCMV-imm+VV), compared with the age-matched control mice (Control+VV), challenged with a lethal dose of VV (_n_=6 mice/group). Mice were infected with VV and the time of death after infection was recorded. Data are representative of three similar experiments.
The decrease in mortality in the LCMV-immune mice was associated with a decreased VV load throughout the course of the acute VV infection (Table 1). By day 3 after infection there was a 50% decrease in VV titers in the lungs of LCMV-immune mice, but by day 5 there were significantly lower VV titers in the spleens and lungs—95% (P<0.05) and 97% (P<0.1), respectively—of the LCMV-immune compared to the control mice. There continued to be a significant 83–90% (P<0.05) reduction in VV titers in the LCMV-immune mice lungs as the VV infection progressed over days 6 and 7. These differences in VV titers were also seen under more stringent conditions in which both groups of mice were depleted of natural killer (NK) cells, as indicated in the two experiments done on day 6 after VV infection (Table 1). These experiments showed that NK cells had not played a role in this protection. The decrease in VV titer was independent of the time of VV challenge after LCMV infection (6–52 weeks, data not shown).
Table 1 Previous immunity to LCMV lowered VV titer during acute VV i.n. challenge
To further examine whether LCMV-specific memory T cells were playing a role in this protective immunity to respiratory infection with VV, LCMV-immune splenocytes were adoptively transferred into naïve mice, which were then challenged with VV. There were significantly lower VV titers in the lungs and pooled MLNs—83% and 99.8%, respectively (P<0.06, _n_=5)—of mice adoptively reconstituted with LCMV-immune splenocytes compared to naïve splenocytes. This showed that the protective heterologous immunity against acute respiratory infection with VV was dependent on splenocytes that contained LCMV-specific memory cells. This finding agreed with published data, which shows that protection depends on both CD8+ and CD4+ T cells in the systemic model17.
LCMV-immune organ-specific CD8+ T cells
To directly examine LCMV epitope–specific memory CD8+ T cells in the lung before and during VV infection and to determine how they could be contributing to the protective immunity, we first defined the LCMV epitope–specific populations present in the lungs, MLNs and spleens of resting LCMV-immune mice. Pooling of data on the intracellular IFN-γ responses to five individual epitopes showed a 16-fold higher percentage of LCMV-specific CD8+ T cells in the lung than in the MLNs (Figs. 2 and 3). The proportion of the CD8+ T cells that were LCMV-specific in the LCMV-immune lung decreased slowly from ∼42% (2.5×105 cells) at 6 weeks to 26% (1.0×105 cells) at 12 weeks and to 14% (9.3×104 cells) at 52 weeks after i.n. LCMV infection. In contrast, the proportion of LCMV-specific CD8+ T cells in the MLNs of these same mice remained stable: 2.2% (2.0×103 cells) at 6 weeks; 1.6% (4.3×103 cells) at 12 weeks; and 2.0% (2.1×103 cells) at 52 weeks; _n_=3 mice/group. The CD8+ memory T cell frequency in the spleen was higher than in the MLNs but was relatively stable after i.n. LCMV infection: 17% (2.0×106 cells) at 13 weeks; 13% (2.1×106 cells) at 15 weeks; 7.3% (1.6×106 cells) at 52 weeks; _n_=3 mice/group. Observations about the stability of memory CD8+ T cells in the spleen after i.p. LCMV infection1,3,33 and differences in the frequency of memory CD8+ T cells in lung, MLN and spleen4,5 were consistent with published data.
Figure 2: Persistence of LCMV epitope–specific memory CD8+ T cells in lungs and MLNs of resting LCMV-immune mice and during i.n. VV infection.
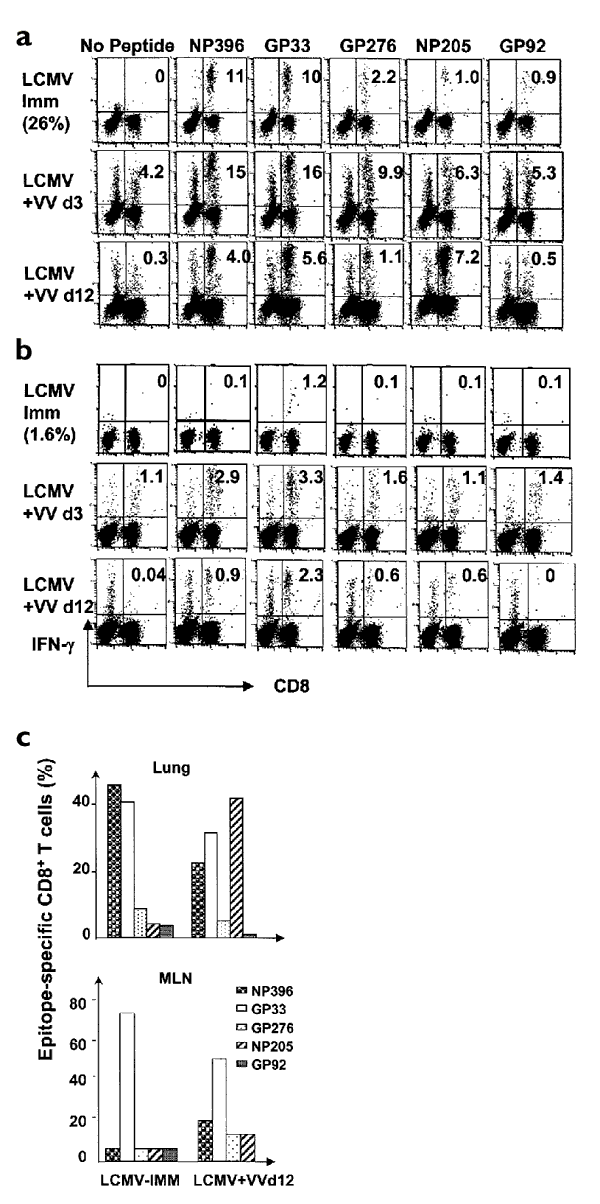
LCMV-immune (>12 weeks after LCMV i.n. infection) and control mice were challenged intranasally with VV. Lymphocytes were isolated from the (a) lungs and (b) MLNs, stimulated with individual peptides and stained for intracellular IFN-γ (see Methods). Lymphocytes from groups of three mice each were pooled before staining. The numbers in the right upper quadrants are the percentages of IFN-γ–staining CD8+ T cells specific for the indicated epitopes. The total LCMV-specific CD8+ T cell frequencies are given in parentheses. IFN-γ staining of cells from the VV-infected control mice was at background amounts (<0.2%). Data are representative of four similar experiments. LCMV Imm, resting LCMV-immune mice; LCMV+VV d3, LCMV-immune mice 3 days after-VV infection; LCMV+VV d12, LCMV-immune mice 12 days after-VV infection. (c) Alterations in the LCMV-specific CD8+ T cell repertoire in the VV-infected LCMV-immune mouse MLN and lung. Data were derived from intracellular the IFN-γ staining analysis in a,b with five known LCMV-specific epitopes and are representative of four similar experiments. LCMV-IMM, resting LCMV-immune mice; LCMV+VVd12, LCMV-immune mice 12 days after-VV infection.
Figure 3: Accumulation of LCMV-specific memory CD8+ T cells in different organs in the LCMV-immune mouse after i.n. challenge with VV.
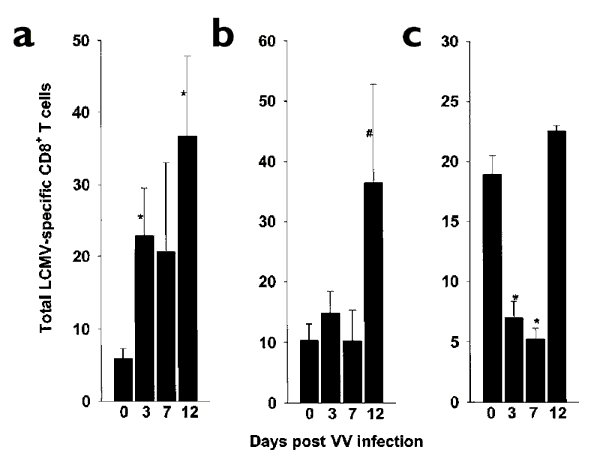
(a) MLN, (b) lung and (c) spleen were examined. The total number of memory CD8+ T cells specific for each LCMV epitope was calculated by multiplying the total cell yield from each mouse organ by the percentage of IFN-γ–staining CD8+ cells specific for that epitope. The sum of the total numbers for each of five LCMV epitopes are shown. Because IFN-γ production by CD8+ T cells without peptide stimulation was observed, this was subtracted from each antigen-specific response before calculating the total numbers. Although some of these CD8+ T cells were LCMV-specific, we used conservative estimates. Statistically significant differences, between the unchallenged and the VV-challenged LCMV-immune mice, in the increase or decrease in total LCMV-specific CD8+ T cells, *P<0.05; #P<0.1. Data represent the mean of four similar experiments. For each experiment, lymphocytes from groups of three mice each were pooled before staining.
Of interest was the unexpected result that the specificity of the CD8+ T cell response between MLN and lung differed. This was because most of the LCMV-specific CD8+ T cells in the MLN were GP33-specific, whereas T cells in the lung had broader specificities, which predominantly targeted NP396 and GP33 with some recognition of GP276 and NP205 (Fig. 2a–c). The spleen tended to have a similar repertoire to the lung; the following results were obtained 13 weeks after LCMV infection. Spleen: GP33, 44%; NP396, 24%; GP276, 11%; NP205, 9%; GP92, 13%. Lung: GP33, 52%; NP396, 27%; GP276, 9%; NP205, 11%; GP92, 1%. These results suggested that different CD8+ memory T cell homeostatic mechanisms were involved in the lung, its draining MLN and the spleen. These differences may have related to recruitment of naïve T cells to the lymph nodes and spleen. Alternatively, once the CD8+ T cells had been recruited to the lung, they may not have easily recirculated. However, neither of these explanations accounts for the difference in repertoire between the lung and MLN.
Organ-specific accumulation of heterologous T cells
After LCMV-immune mice were acutely challenged with VV, LCMV epitope-specific CD8+ memory T cells in both the lungs and MLNs were not diluted out, as one might have expected, by the VV-specific CD8+ T cell response (Figs. 2a,b and 3). In fact, the total number of LCMV-specific CD8+ T cells in the MLN increased fourfold 3 days after VV challenge and continued to rise to sixfold higher by day 12 after infection (Fig. 3a). There was only a moderate increase in LCMV-specific CD8+ T cells in the lung 3 days after VV challenge, but by day 12 after infection there was a fourfold increase (Fig. 3b). In contrast to these results, the spleen showed a significant 67% decrease (P<0.05) of LCMV-specific CD8+ T cells by day 3 and a further decrease by day 7. Then, by day 12 after infection, the number of LCMV-specific CD8+ T cells returned to the numbers in resting LCMV-immune mice (Fig. 3c). All these changes occurred in the presence of VV antigens, as VV titers in lungs sampled in one of two representative experiments stabilized from day 3 of i.n. challenge until day 9; titers then declined in both control and LCMV-immune mice. (Representative data from control mice: day 3, 6.4±0.02 log PFU/lung; day 7, 6.8±0.2 log PFU/lung; day 9, 6.1±0.5 log PFU/lung; day 13, 3.0±0.5 log PFU/lung; _n_=2 mice/group). By day 15, VV could not be detected in the lung homogenates.
Modulation of organ-specific heterologous T cells
To examine whether there was a VV-induced nonspecific bystander accumulation of all LCMV-specific CD8+ T cells that was equal in the lungs and MLNs or whether there was some selectivity to this accumulation, we analyzed the proportion each epitope-specific CD8+ T cell population represented of the total LCMV-specific pool (Fig. 2c). As already noted, there was a different LCMV-specific repertoire in the resting LCMV-immune MLN and lung: GP33-specific CD8+ T cells dominated the repertoire in the MLN, whereas NP396- and GP33-specific CD8+ T cells codominated in the lung. We found that VV infection modified the LCMV-specific repertoire differently in both these two organs in such a manner that it further enhanced the difference between these two sites. In the MLN, by day 12 after VV, there was a significant (P<0.05) and consistent skewing of the LCMV repertoire in all four experiments, which resulted in increased proportions of NP396- and GP276-specific CD8+ T cells and a decreased proportion of GP33-specific CD8+ T cells, respectively (Fig. 2c). In the lung there was a different skewing of the LCMV-specific repertoire and, although between experiments there was more variability in the lung than in the MLN, there was always a marked change in the selective specificities with either the GP33- or NP205-specific responses that dominated the repertoire. In one representative experiment (Fig. 2c), there was a sevenfold selective expansion of the NP205-specific population, retention of the GP33-specific population, but a 50–67% loss of the NP396- and GP276-specific populations. Overall, these data indicated that VV infection in an LCMV immune mouse can markedly alter, in an organ-dependent manner, the LCMV-specific memory T cell population.
Production of IFN-γ in vivo
We questioned whether there was any direct evidence that these accumulating LCMV-specific memory CD8+ T cells were activated in vivo during VV infection. Three days after VV infection, 4.2% of CD8+ T cells produced IFN-γ after incubation in vitro without peptide stimulation (Fig. 2a). This suggested that CD8+ T cells had been activated in vivo during the VV infection to produce IFN-γ. To determine whether these CD8+ T cells were LCMV-specific memory cells activated in vivo, we did a modified intracellular IFN-γ assay. Immediately after isolation, the lung lymphocytes, without any in vitro stimulation or incubation, were stained on ice for intracellular IFN-γ and with major histocompatibility complex (MHC) dimers specific for three of six known LCMV epitopes (Fig. 4a). There was no production of IFN-γ by LCMV-epitope specific memory CD8+ T cells isolated from resting LCMV-immune mice. Compared to the minimal in vivo IFN-γ production of 0.7% by CD8+ T cells (4.2×102 cells) from the VV-infected control mouse lungs, 16% of CD8+ T cells (6.2×104 cells) from the LCMV-immune mouse lungs produced IFN-γ in vivo 3 days after VV challenge. Among them, 30% of the NP205-specific, 18% of the GP33-specific and 16% of the NP396-specific CD8+ memory T cells in the lung produced IFN-γ. These three LCMV epitope-specific populations accounted for 4.1% of the CD8+ T cells that produced IFN-γ in vivo in response to VV.
Figure 4: Activation of LCMV-specific CD8+ T cells in mouse lungs early on after VV infection.
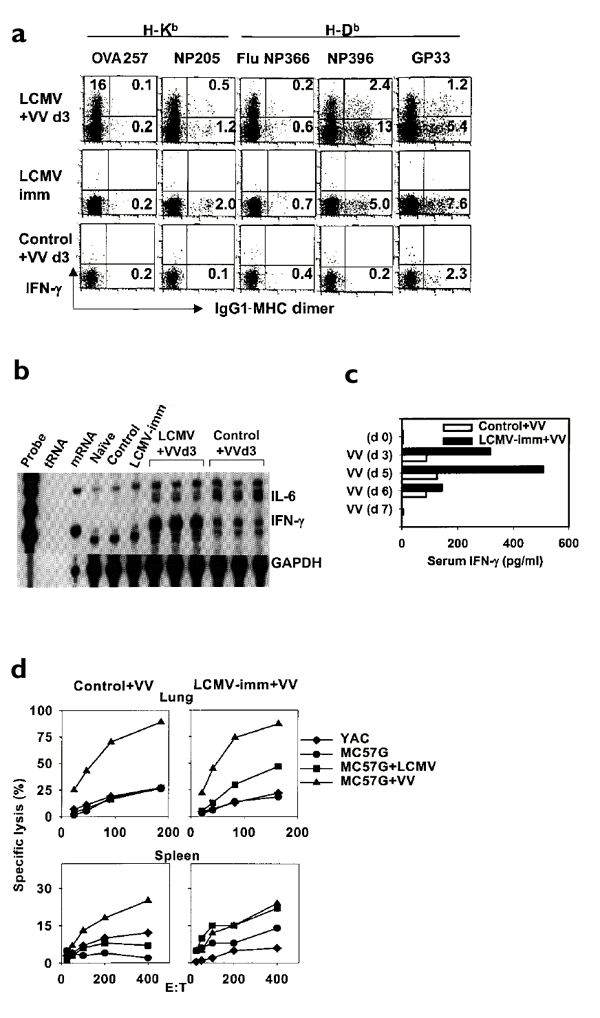
(a) Increased in vivo IFN-γ production by LCMV-specific CD8+ memory T cells after VV infection. Lymphocytes isolated from mouse lungs were pooled and immediately stained with anti-CD8+, LCMV epitope–specific MHC dimers and anti–IFN-γ without in vitro stimulation. Data were gated on CD8+ T cells. Increased IFN-γ in LCMV-immune mice after VV as assessed by (b) an RNase protection assay of cytokine mRNA levels in the lung tissue and (c) an ELISA assay that measured IFN-γ in pooled sera. tRNA, negative control; mRNA, positive control; GAPDH, the housekeeping gene (used to normalize mRNA levels). (d) Activation of LCMV-specific cytotoxicity in pooled lymphocytes isolated from the lungs and spleens of LCMV-immune and control mice 6 days after VV infection. Mice were injected with anti-NK1.1 to deplete NK cells. Data for each experiment were derived from lymphocytes pooled from three mice/group and are representative of three similar experiments. E:T, effector to target ratio.
This experiment with MHC dimers charged with three of the six LCMV-specific epitopes accounted for one-quarter of the IFN-γ–producing cells being LCMV-specific. Two additional similar experiments with MHC dimers to define four of the LCMV epitope–specific responses (NP205, NP396, GP33 and GP34) showed that 11% and 10% of the lung CD8+ T cells produced intracellular IFN-γ 3 days after VV infection. In addition, 58% and 42% of those responses, respectively, could be attributed to CD8+ T cells specific to those LCMV epitopes. These experiments probably indicated that, at this time after VV infection, well over half of the IFN-γ–producing cells were LCMV-specific. This was because not all the LCMV epitopes are defined, two of the immunodominant epitopes were not tested and it is unlikely that low-affinity responses or responses by receptor down-modulated T cells would be recorded. This, of course does not rule out the possibility that some of this response was mediated by T cells not specific to LCMV but was perhaps activated in the presence of the LCMV-specific T cells.
The control H-Kb–dimer loaded with ovalbumin-specific peptide, OVA(257–264), bound <0.2% of CD8+ T cells. The control H-Db–dimer loaded with the influenza peptide, NP(366–374), bound <0.8% of CD8+ T cells; these cells produced minimal IFN-γ. Some degree of recognition of influenza NP(366–374)-pulsed targets by LCMV-specific CTLs has been observed10. The low degree of binding of the GP33-specific dimers to CD8+ T cells from the lungs of VV-infected control mice was observed by CD8+ T cells from the lungs of noninfected control mice (data not shown), which suggested that GP33 might cross-react with VV and some self antigens. NK cells (16%, 2.3×104 cells), CD4+ T cells (0.2%, 1.0×103 cells) and γδ T cells (2.6%, 6.0×102 cells) pooled from three VV-infected LCMV-immune mice also produced IFN-γ in vivo. However, there was no increase in IFN-γ production by these cells in LCMV-immune mice 3 days after VV challenge—which is considered the peak of the response by NK and γδ T cells activity34,35—compared to the VV-infected control mice (lung lymphocytes pooled from three mice: NK cells, 23%, 3.7×104 cells; CD4+ T cells, 0.5%, 1.4×103 cells; and γδ T cells, 1.7%, 4.0×102 cells). In addition, the CD8+ T cells that produced IFN-γ in vivo did not express the NK cell–associated molecule NK1.1.
Consistent with the above observations, RNase protection assays showed 4.7-fold higher levels of IFN-γ mRNA in the LCMV-immune mouse lungs, compared to VV-challenged control mouse lungs, 3 days after VV challenge (relative intensity of IFN-γ: LCMV+VV, 0.33±0.01; control +VV, 0.07±0.003, P<0.00003) (Fig. 4b). In the unchallenged LCMV-immune mouse lungs, compared to the nonchallenged control mouse lungs, a low basal amount of IFN-γ mRNA was observed. In addition, three- to fivefold higher concentrations of IFN-γ protein—as detected by enzyme linked immunosorbant assay (ELISA)—were observed in the sera of VV-challenged LCMV-immune mice (Fig. 4c). This began at day 3 and the greatest difference occurred by day 5 after VV. In contrast, after VV challenge twofold higher mRNA levels of IL-6, a potent proinflammatory cytokine, were observed in control mouse lungs compared to the LCMV-immune mouse lungs (relative intensity of IL-6: LCMV+VV, 0.04±0.0003; control +VV, 0.08±0.01, P<0.05). (Fig. 4b). Higher amounts of serum IL-6 were also found in the VV-infected control mice (42 pg/ml, sera pooled from three mice) compared to the VV-challenged LCMV-immune mice (12 pg/ml, sera pooled from three mice). This suggested that a stronger acute inflammatory response took place in the control mice and that previous immunity to LCMV markedly altered the cytokine responses induced by VV infection.
Because IFN-γ has marked impact on VV replication36, to further test whether the increased IFN-γ produced in the LCMV-immune mice was important in this protective immunity, LCMV-immune mice were treated with anti–IFN-γ and infected with VV. In one of the two similar experiments, compared to control monoclonal antibody (mAb) treatment, anti–IFN-γ treatment resulted in 3.2-fold higher VV titers in the lungs (anti–IFN-γ: 6.8±0.1 log PFU; control IgG: 6.3±0.1 log PFU; P<0.02; _n_=5) and 16-fold higher titers in the pooled MLNs (anti–IFN-γ: 2.7 log PFU; control IgG: 1.5 log PFU, _n_=5) of LCMV-immune mice. The importance of IFN-γ in regulating VV synthesis in LCMV-immune mice has been documented in mice during systemic immunity17.
Cytolytic function in heterologous T cells
We further assessed the activation status of the accumulating LCMV-specific memory CD8+ T cells by determining their cytolytic capacity ex vivo in bulk cytotoxicity assays of pooled mouse lung lymphocytes and splenocytes (Fig. 4d). Both LCMV-immune and control mice that had been treated with anti-NK1.1 to deplete NK cells in vivo, mounted equivalent VV-specific CTL responses on day 6 after VV challenge. However, only in T cells from the VV-infected LCMV-immune mice did a VV-induced reactivation of LCMV-specific cytotoxicity occur (Fig. 4d). In nonchallenged LCMV-immune and control mice, there was no LCMV-specific lysis (data not shown). These results further supported the findings that LCMV-specific CD8+ T cells were not only accumulating but were also functionally activated during acute VV infection.
Lymphocyte-dependent pathology
To examine whether immunity to LCMV would influence VV-induced pathology in the lung, tissue sections were stained with hematoxylin and eosin (Fig. 5). Compared to sections from naïve mouse lungs (Fig. 5a), sections from LCMV-immune mouse lungs (Fig. 5b) showed some mononuclear (MN) infiltrating cells and occasional lymphocyte aggregates in the interstitium, a pattern that can be seen in older naïve mice in any clean mouse colony. The majority of the LCMV-immune lung architecture was normal.
Figure 5: Enhanced lymphocytic infiltration in the lungs of LCMV-immune mice after infection with VV and the role of IFN-γ.
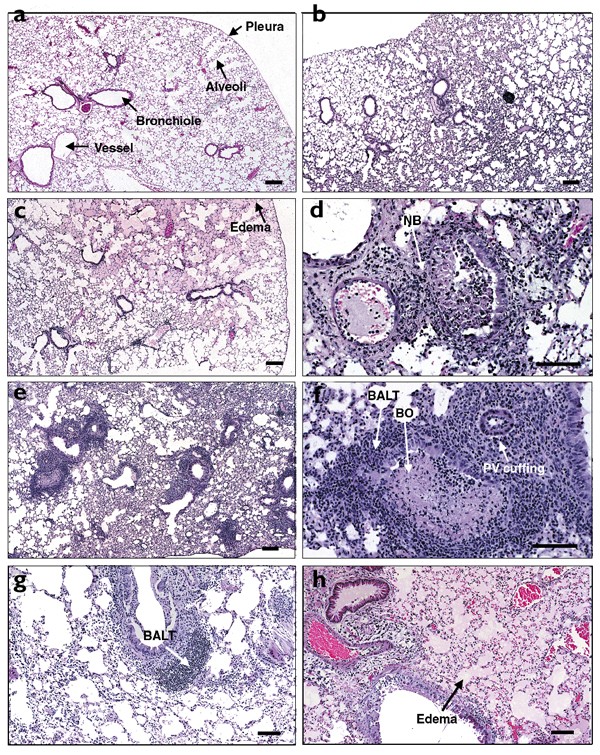
Lung sections from mice that did not receive VV (a,b) or had been infected with VV 7 days previously (c–h) were stained with hematoxylin and eosin. (a) Naïve mouse lungs showed normal architecture. (b) LCMV-immune mouse lungs had mild lymphocytic infiltrates in the interstitium. (c) VV-infected control mouse lungs mainly showed severe alveolar edema (pink material in air spaces) and (d) acute necrotizing bronchiolitis (NB) and peribronchiolitis as well as neutrophilic infiltrates in the interstitium. (e) VV-infected LCMV-immune mouse lungs had greatly increased BALT around the airways and no alveolar edema. (f) BALT surrounded an airway with bronchiolitis obliterans (BO) and perivascular (PV) lymphocytic cuffing was also present. (g) VV-infected LCMV-immune mice were treated with control IgG1. (h) In vivo depletion of IFN-γ in VV-infected LCMV-immune mice by daily i.v. and i.p. injection of anti–IFN-γ for 6 days resulted in severe pulmonary edema and no induction of BALT. Bars, 100 μm.
Seven days after VV infection, the lungs from the LCMV-immune and control mice had completely different pathologies (Fig. 5c,e and Table 2). We found that 72% of the control mouse lungs had severe alveolar edema (Fig. 5c and Table 2): an accumulation of the extravascular fluid in the air spaces, which presumably disturbed gas exchange. In addition, the VV-infected control mouse lungs mainly showed acute mixed inflammatory infiltrates (AMI) with polymorphonuclear (PMN) and MN cells in the peribronchial areas (86%) and interstitium (71%), as well as in the perivascular areas (71%) (Fig. 5d and Table 2). In contrast, the lungs of LCMV-immune mice challenged with VV showed a prominent lymphocytic response, which may have aided in the clearance of VV. In addition, 80% of the LCMV-immune mouse lungs had marked induction of the lung lymphoid system (Fig. 5e and Table 2), as shown by prominent bronchus-associated lymphoid tissue (BALT). BALT was defined by nodules of lymphoid tissue in the bronchial lamina propria near the branch points of an airway or between the bronchus and an artery18. Although acute LCMV infection induced a minimal transient BALT, there was no BALT present in the resting LCMV-immune mouse lungs (Fig. 5b). Prominent chronic MN infiltration (CMI) with lymphocytes and macrophages was present in the interstitium and pleura of almost all the VV-infected LCMV-immune mouse lungs.
Table 2 Altered immunopathology in the intranasally VV-infected LCMV-immune mouse lungs
In addition, 60% of the LCMV-immune lungs had perivascular lymphocytic cuffing, as defined by lymphocyte accumulation around the vessels (Fig. 5f and Table 2). LCMV-immune mouse lungs challenged with VV also showed the less common pathology of necrotizing vasculitis (60%) and bronchiolitis obliterans (40%) (Fig. 5f), which, in humans, are thought to be immune-mediated diseases of unknown etiology. By 7 days after VV infection, lungs from both groups showed necrotizing bronchiolitis, one of the main lesions of the disease (Fig. 5d,f), as defined by the presence of necrotic bronchiolar epithelium with PMNs and debris. Thus, altered immunopathological features were seen in the VV-infected LCMV-immune mice lungs, which showed a prominent lymphocytic response, whereas the VV-infected control mouse lungs mainly had acute neutrophilic infiltrates and severe edema.
Because the LCMV-specific memory CD8+ T cells produced large amounts of IFN-γ in the lung early in VV infection, we questioned whether IFN-γ played a role in the altered lung pathology after VV challenge. Compared to the VV-infected LCMV-immune mice treated with control IgG1 (Fig. 5g), in vivo depletion of IFN-γ in the VV-infected LCMV-immune mice by intravenous (i.v.) and i.p. injection of anti–IFN-γ resulted in severe edema and no induction of BALT (Fig. 5h). This pathology was similar to the changes observed in the VV-infected control mouse lungs (Fig. 5c), which suggested that IFN-γ might play a role in recruiting the lymphocytes to the lungs and thus alter the pathology in the VV-infected LCMV-immune mouse lungs. In support of this, IFN-γ participates in inducing chemokines to recruit activated lymphocytes to the site of viral replication37.
LCMV-specific CD8+ T cells in BALT
To determine whether the BALT in the lungs of LCMV-immune mice acutely infected with VV was composed of LCMV-specific memory CD8+ T cells, we stained lung sections in situ with MHC tetramers, a technique that locates antigen-specific T cells in tissues38,39. We chose NP396-specific tetramers for the staining of LCMV-specific CD8+ T cells, as NP396-specific T cells bind to their antigen at 1000-fold higher affinity than other LCMV epitope-specific T cells40. Because BALT was not prominent in VV-infected control mouse lungs, compared to the LCMV-immune lungs—which had prominent BALT (Fig. 6a)—there were few clusters of CD8+ T cells in those lungs (Fig. 6b). The few CD8+ T cells that were present in the VV-infected control mouse lungs did not costain with NP396-tetramer (Fig. 6b). In the LCMV-immune lungs there were many CD8+ T cells that costained with NP396-tetramer after VV challenge (Fig. 6a). This showed that LCMV-specific CD8+ T cells were recruited and contributed to the formation of the prominent BALT, one of the hallmarks of the altered pathology present in these mice.
Figure 6: In situ detection of LCMV epitope–specific CD8+ T cells in lung tissue with NP396-specific tetramer.
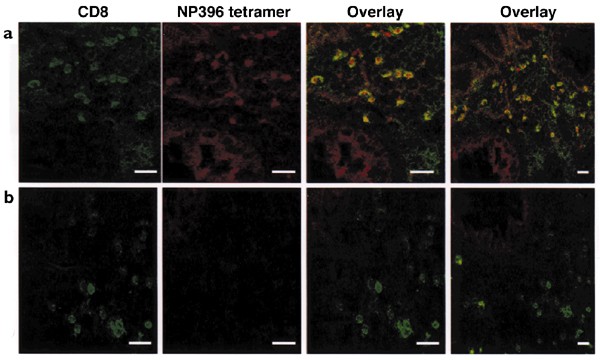
Seven days after VV challenge, lungs from (a) LCMV-immune and (b) control mice were collected. Frozen sections (20 μm) were cut and stained. Results are shown as single color with anti-CD8+ (green), NP396–MHC tetramer (red) and colored overlay (yellow) of these two stains. The right-hand panels in a and b show a larger area of lung tissue. Data are representative of three similar experiments. Bars, 25 μm.
Discussion
This study systematically examined what effect, if any, heterologous virus challenge had on complex memory CD8+ T cell populations. We showed, in a respiratory infection model, that a second unrelated viral infection can have a profound impact on memory T cells specific to a previously encountered virus. This impact resulted in: (i) the activation of cytotoxic function and IFN-γ production of these memory T cells; (ii) the selective expansion and modulation of their original T cell repertoire; and (iii) the selective organ-dependent compartmental redistribution of these defined antigen-specific heterologous memory T cell populations. As a consequence, these responses affected the course of the second infection, which resulted in enhanced protective immunity and greatly altered immunopathology.
The most significant evidence that memory CD8+ T cells specific to LCMV were activated in vivo by VV to produce IFN-γ was provided by the intracellular staining of lymphocytes immediately after isolation from the lungs of day 3 VV-infected LCMV-immune mice that had not received any form of in vitro stimulation. IFN-γ has a marked impact on VV replication36. Increased IFN-γ production appeared to play a role both in decreasing the virus load and altering the immunopathology, as anti–IFN-γ enhanced VV titers in LCMV-immune mice and blocked the infiltration of the lung with LCMV-specific memory T cells upon VV challenge. Although the lung is not considered to be a conventional lymphoid organ, it does have a prominent immune system that consists of the intravascular leukocyte pool and the leukocytes in the interstitium, the bronchoalveolar spaces and the BALT. In humans and mice BALT is not a constitutive structure of the lung, but it can be initiated and vigorously expanded upon infection to become a highly integrated mucosal immune system along the respiratory tract18. By histological examination and in situ tetramer staining we showed that the accumulation of LCMV-specific memory CD8+ T cells in the BALT contributed to a marked change in the architecture of the lung during VV infection. These changes were ablated by treatment with anti–IFN-γ. The absence of these activated memory T cells resulted in severe acute inflammatory responses and associated fulminant pulmonary edema that may be one of the factors that contributes to the high mortality of control mice upon VV infection.
The presence of these activated memory T cells may also have contributed to some of the VV-infected LCMV-immune mice developing bronchiolitis obliterans, which is an obstruction of bronchioles by plugs of fibrin and inflammatory cells. In humans this is a condition whose etiology is not well understood; it is much like panniculitis, which can be observed in the fat pads of LCMV-immune mice challenged with VV during i.p. infections17. Both of these disease processes in humans are probably immune-mediated, occurring in some viral and intracellular bacterial infections or in response to some drugs17,41. This mouse model suggests that an individual's past history of infections may influence whether they develop bronchiolitis obliterans or panniculitis (erythema nodosum) in response to an additional antigenic challenge. Studying this type of model might provide further insights into these poorly understood conditions.
Not only was there a functional activation of LCMV-specific memory CD8+ T cells, there was a continued selective expansion and accumulation of certain epitope-specific populations in the presence of VV antigen. Although it was highly likely that the chemokines and cytokines produced during VV infection assisted in the initial recruitment of LCMV-specific memory CD8+ T cells from the spleen into the lung and MLN, they were unlikely to account for the selective epitope-specific nature of this expansion. We believe that cross-reactive T cell responses likely participated in their activation and selective expansion in the lung. Certain T cell clones recognize both LCMV- and VV-infected targets12, and bulk CTL analyses with VV-infected LCMV-immune mice indicated that GP33-coated targets—in comparison to NP396- or GP276-coated targets—are preferentially lysed (reactivity with NP205 was not examined)22. Three VV peptides with partial homology to LCMV NP205 have been identified; they all can stimulate LCMV-specific CD8+ T cells to produce IFN-γ (L. K. Selin et al., unpublished data). Another study suggested that VV could induce bystander activation of LCMV GP33-transgenic T cells42, but cross-reactivity was not ruled out. In addition, other studies found very little evidence for LCMV causing expansion of naïve or memory transgenic T cells that did not cross-react with LCMV33,43.
These studies support the concept that there is remarkable complexity and plasticity in CD8+ memory T cell responses. Memory CD8+ T cells in a resting immune host exist in distinct populations distinguished by activation state7,10, the expression of cell surface molecules such as chemokine and cytokine receptors6,9,44,45 and their distribution frequencies in different organs4,5. It appears from our results that different organs have different T cell repertoires. This observation is important as it further supports the concept that memory CD8+ T cell pools are not homogeneous throughout the host and are not equal in all sites. We found that challenge with the second virus resulted in a rapid accumulation of LCMV-specific memory CD8+ T cells in the MLN, where the frequency had been relatively low, with a more gradual increase in the lung where the frequency was already very high. As these memory CD8+ T cells accumulated in these two sites, their repertoire became further skewed, with selective expansion of specific epitopes; once again each site developed a different repertoire. It is not clear what other mechanisms drive this qualitative and quantitative difference in CD8+ memory T cells in different sites. Memory T cells can be divided into at least two functional types based on chemokine receptor and homing receptor expression44. It is possible that the functional type of each LCMV epitope–specific memory T cell population may influence where they traffic during the resting memory state and during acute VV challenge.
Heterologous immunity mediated by memory T cells is a new research area that may clarify our understanding of how memory T cells function in immunity and pathogenesis in the natural environment as the host is exposed to multiple different pathogens over a lifetime. Heterologous immunity may be an under-appreciated aspect of host responses to infection in general. Many viruses in humans—such as Epstein-Barr virus, varicella-zoster and the 1918 influenza virus strain—cause much more severe infections in teenagers and young adults than they do in younger children46,47,48. Could such a difference be due to immunopathology occurring as a consequence of the reactivation of memory cells, which may be more diverse and prominent in a more immunologically mature individual? Several studies have begun to scratch the surface of this complex but poorly understood phenomenon. For instance, in the search for HIV vaccines, researchers have shown that a secondary challenge with a VV recombinant expressing an HIV epitope can greatly enhance the immune response to HIV DNA vaccination49,50. But what are the mechanisms driving this? There is an inverse correlation between early exposure of humans to Bacillus Calmette-Guerin (BCG) vaccination and the development of atopic disorders51. In mice, previous exposure to BCG, a strong T cell and IFN-γ stimulus, was correlated with suppression of development of local inflammatory T helper type 2 (TH2) responses and allergen-induced eosinophilia in the lung52. A reactivation of memory T cells in an otherwise poorly protective immune response may have contributed to the severity of the infections in children receiving the 1962 respiratory syncytial virus (RSV) vaccine53. In animal models, vaccination with recombinant VVGPRSV (rVVGPRSV) shortly after acute influenza infection greatly reduces the TH2-like eosinophilia response that occurs upon challenge, 5 weeks later, with live RSV54. In addition, dengue virus–immune individuals infected with a second dengue serotype suffer severe immunopathological responses. These responses may occur as a consequence of potent reactivation of cross-reactive T cells in the absence of protective antibody responses55. Some of these examples argue that in the absence of a completely protective immune response that occurs with homologous infection, the reactivation of memory T cells by heterologous agents may cause immunopathological damage. Alternatively, as we have shown here, the reactivation of heterologous memory T cells may also contribute to a protective response. The lung may have a strong predilection for these effects, as memory CD8+ T cells are present at such high frequency for prolonged periods.
Methods
Virus infections of mice.
Five-week-old C57BL/6 (H-2b) male mice were from The Jackson Laboratory (Bar Harbor, ME). The mice were anesthetized by inhalation of metofane (Pitman-Moore, Mundelein, IL) and infected intranasally with 4×105 PFU of the Armstrong strain of LCMV propagated in baby hamster kidney cells (BHK21)12. Other mice were inoculated with BHK21 cell culture supernatant as controls. After the acute T cell response had been stored and the immune system had returned to homeostasis (6 weeks or longer), the immune and control mice were challenged intranasally with the WR strain of VV propagated in L929 cells12 and purified from serum contaminants by centrifugation on sucrose density gradients. Because of some variability of virulence among different VV stocks, the VV dose varied from 104 to 105 PFU. All the experiments, except for the mortality study (Fig. 1), were done with sublethal doses of VV. In the mortality study, for a specific virus stock we used a dose that killed 60–80% of naïve nonimmune C57BL/6 mice. The immune mice were age-matched to the control mice and housed under exactly the same pathogen-free conditions for the same time period. All experiments were done in compliance with institutional guidelines as approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School.
Virus titration.
The number of VV PFU in each organ (MLN, spleen and lung) was determined by plaque assays with the use of a 10% homogenate of tissue taken from individual mice, as described17.
Adoptive cell transfer.
Naïve C57BL/6 mice were injected intravenously with one spleen equivalent (∼5×107–10×107 cells) of naïve splenocytes or LCMV-immune splenocytes and challenged intranasally with VV. On 6.5 day after infection, lungs, MLNs and spleens were collected and titrated for VV.
Isolation of lung lymphocytes.
Lung lymphocytes were isolated using an adaptation of a previously described protocol56. The lung vascular bed was first flushed with 10 ml of chilled Hanks' balanced salt solution (HBSS, Gibco-BRL, Gaithersburg, MD) introduced via cannulation of the right ventricle of the heart. The excised lungs, separated from all the associated lymph nodes, were minced and incubated for 1 h at 37 °C in 5 ml of HBSS (10% fetal calf serum (FBS)) containing 125 U/ml of collagenase I (Gibco-BRL), 60 U/ml of DNase I and 60 U/ml of hyaluronidase (Sigma, St. Louis, MO). The resulting single cell suspension was spread over Lympholyte-M (Cedarlane Labs, Hornby, Canada) and centrifuged at 500_g_ for 20 min at 25 °C. Then the lymphocytes were collected.
Peptide-induced intracellular IFN-γ staining.
Based on the protocol from Cytofix/Cytoperm Plug with Golgiplug Kit (PharMingen, San Diego, CA), the isolated cells were stimulated with peptide (5 μg/ml) in the presence of 10 U/ml of human recombinant IL-2 and 0.2 μl of Golgiplug for 5 h at 37 °C. The cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD8+ (clone 53-6.7), then fixed and permeabilized with Cytofix/Cytoperm Plug solution. They were then stained with phycoerythrin (PE)-conjugated rat anti–IFN-γ (PharMingen) or control PE-conjugated rat IgG1 isotype. The cells were analyzed either on a FACS 440 or FACSstar plus (Becton Dickinson, San Jose, CA). For each experiment, we pooled lymphocytes from three mice because we wished to sample a large number of mice in experiments. This involved large numbers of samples and, at the same time, we examined responses to five peptides plus isotype controls at four different time-points in three organs. In addition, we could not obtain enough lymphocytes to test five peptides from the lung or MLN of one mouse.
H-Db– and H-Kb–IgG1 MHC dimer staining and modified intracellular staining for in vivo IFN-γ.
Immediately after isolation from mouse lungs, lymphocytes were stained with a mouse H-Db– or H-Kb–IgG1 fusion protein conjugated to β2-microglobulin and the appropriate peptide22,23. Briefly, 1 μg of the peptide–H-Db–IgG1 or peptide–H-Kb–IgG1 MHC dimer mixture was incubated with cells for 1.5 h at 4 °C. The cells were then stained with FITC–anti-CD8+ (clone 53-6.7) and biotinylated anti–mouse IgG1 (PharMingen) for 30 min at 4 °C, followed by incubation with PE-streptavidin (PharMingen) for 20 min at 4 °C. The cells were then fixed and permeabilized with Cytofix/Cytoperm Plug solution. They were stained with allophycocyanin-conjugated anti–IFN-γ (clone R4-6A2, PharMingen) or control allophycocyanin-conjugated IgG1 isotype. As no in vitro stimulation or incubation was carried out, any positive staining for intracellular IFN-γ indicated that the CD8+ T cells producing IFN-γ would have been directly activated in vivo by the infection. All staining with the isotype control was consistently <0.04%. A second control was also done using purified anti–IFN-γ (clone R4-6A2, PharMingen), which specifically blocked intracellular IFN-γ staining with allophycocyanin-conjugated anti–IFN-γ. The cells were analyzed either on a FACS 440 or FACSstar plus (Becton Dickinson).
Cytokine RNase protection assay.
Total RNA was extracted from the whole lung tissue with Trizol agent (Gibco-BRL). Detection and quantification of a variety of murine cytokine mRNAs was done with the multiprobe RNase protection assay system (PharMingen). Briefly, a mixture of [γ-32P]UTP-labeled antisense riboprobes was generated from a panel of different cytokine templates plus the template for the control murine housekeeping gene encoding glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The RNA sample (20–40 μg) was hybridized overnight at 56 °C with 32P-labelled riboprobe mixture, then digested with RNase. RNase-protected RNA fragments were purified and then resolved on 5% PAGE. The protected bands were observed after exposure of the gels to phosphor imager and X-ray films and quantified by densitometric analysis with Image Quant densitometric software.
Cytokine ELISA assay.
Serum concentrations of IFN-γ and IL-6 were measured by ELISA, as per PharMingen's protocol. The sera isolated from the blood of LCMV-immune and control mice 3, 5, 6 and 7 days after acute i.n. VV challenge was analyzed by a PharMingen mouse IFN-γ ELISA assay, as per the manufacturer's protocol. Sera from groups of three mice each were pooled before ELISA.
In vivo IFN-γ depletion.
In vivo depletion of IFN-γ was done daily by i.v. (days 0 and 3) and i.p. injection of 100 μl of ascites of rat anti–IFN-γ (ATCC R4-6A2) or a control rat IgG1 isotype (clone A110-1, PharMingen) after VV challenge. At day 6.5 after VV infection, the lungs, MLNs and spleens were collected and underwent virus titration and histological analysis.
Direct ex vivo cytotoxicity assay.
Cell-mediated cytotoxicity was determined with a standard 51Cr-release CTL assay, as described10. 51Cr-labeled YAC cells were used as NK cell targets. 51Cr-labeled MC57G cells, either uninfected or infected with LCMV or VV at a multiplicity of infection of 0.1 and 10 PFU/cell, respectively, were used as targets for CTL.
Lung histological evaluation.
At various time-points 3–30 days after VV challenge, lungs from LCMV-immune and control mice were collected, fixed in 10% neutral buffered formaldehyde and then paraffin-embedded. Tissue sections (5 μm) were stained with hematoxylin and eosin and analyzed microscopically. Scores for lung pathology were graded concurrently by two pathologists who were blinded in regards to treatments the mice had received.
In situ MHC-tetramer staining and confocal laser scanning microscopy (CLSM) of lung tissue sections.
NP396-specific H-Db tetramers were prepared as described57. Lungs from the control and LCMV-immune mice were frozen in OCT compound (Miles Co., Eikhart, IN). Sections (20 μm) were cut, mounted on microscope slides and blocked by purified 2.4G2 antibody (PharMingen) specific for Fc receptors for 15 min at 4 °C, then incubated overnight at 4°C with FITC–anti-CD8+ (6 μg/ml, clone 53-5.8, PharMingen) and PE-labeled NP396-specific MHC tetramer (20 μg/ml) in PBS–10% normal mouse serum, followed by washing three times with PBS–10% normal mouse serum38. Sections were protected by The ProLongTM antifade kit (Molecular Probe, Eugene, OR) before CLSM. Green (FITC) and red (PE) signals were collected on a Leica TCS NT (Leica Microsystems, Heidelberg, Germany) confocal system. Images were taken with a 40× NA1.4 objective. Possible overlapping signals between the different fluorochromes were subtracted.
Statistical analysis.
The Student's _t_-test was used for data analysis.
References
- Murali-Krishna, K. et al. Counting antigen-specific CD8+ T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187 (1998).
Article CAS Google Scholar - Flynn, K. J. et al. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity 8, 683–691 (1998).
Article CAS Google Scholar - Selin, L. K., Vergilis, K., Welsh, R. M. & Nahill, S. R. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J. Exp. Med. 183, 2489–2499 (1996).
Article CAS Google Scholar - Hogan, R. J. et al. Activated antigen-specific CD8+ T Cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 166, 1813–1822 (2001).
Article CAS Google Scholar - Masopust, D., Vezys, V., Marzo, A. L. & Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001).
Article CAS Google Scholar - Razvi, E. S., Welsh, R. M. & McFarland, H. I. In vivo state of antiviral CTL precursors. Characterization of a cycling cell population containing CTL precursors in immune mice. J. Immunol. 154, 620–632 (1995).
CAS PubMed Google Scholar - Tabi, Z., Lynch, F., Ceredig, R., Allan, J. E. & Doherty, P. C. Virus-specific memory T cells are Pgp-1+ and can be selectively activated with phorbol ester and calcium ionophore. Cell Immunol. 113, 268–277 (1988).
Article CAS Google Scholar - Tough, D. F., Sun, S., Zhang, X. & Sprent, J. Stimulation of naïve and memory T cells by cytokines. Immunol. Rev. 170, 39–47 (1999).
Article CAS Google Scholar - Bradley, L. M., Croft, M. & Swain, S. L. T-cell memory: new perspectives. Immunol. Today 14, 197–199 (1993).
Article CAS Google Scholar - Selin, L. K. & Welsh, R. M. Cytolytically active memory CTL present in lymphocytic choriomeningitis virus-immune mice after clearance of virus infection. J. Immunol. 158, 5366–5373 (1997).
CAS PubMed Google Scholar - Yang, H. Y., Dundon, P. L., Nahill, S. R. & Welsh, R. M. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J. Immunol. 142, 1710–1718 (1989).
CAS PubMed Google Scholar - Selin, L. K., Nahill, S. R. & Welsh, R. M. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 179, 1933–1943 (1994).
Article CAS Google Scholar - Kuwano, K., Reyes, V. E., Humphreys, R. E. & Ennis, F. A. Recognition of disparate HA and NS1 peptides by an H-2Kd-restricted, influenza specific CTL clone. Mol. Immunol. 28, 1–7 (1991).
Article CAS Google Scholar - Shimojo, N., Maloy, W. L., Anderson, R. W., Biddison, W. E. & Coligan, J. E. Specificity of peptide binding by the HLA-A2.1 molecule. J. Immunol. 143, 2939–2947 (1989).
CAS PubMed Google Scholar - Loftus, D. J., Chen, Y., Covell, D. G., Engelhard, V. H. & Appella, E. Differential contact of disparate class I/peptide complexes as the basis for epitope cross-recognition by a single T cell receptor. J. Immunol. 158, 3651–3658 (1997).
CAS PubMed Google Scholar - Mason, D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404 (1998).
Article CAS Google Scholar - Selin, L. K., Varga, S. M., Wong, I. C. & Welsh, R. M. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J. Exp. Med. 188, 1705–1715 (1998).
Article CAS Google Scholar - Pabst, R. Is BALT a major component of the human lung immune system? Immunol. Today 13, 119–122 (1992).
Article CAS Google Scholar - Elson, C. O. In defense of mucosal surfaces. Regulation and manipulation of the mucosal immune system. Adv. Exp. Med. Biol. 412, 373–385 (1997).
Article CAS Google Scholar - Altman, J. D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996).
Article CAS Google Scholar - Callan, M. F. et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J. Exp. Med. 187, 1395–1402 (1998).
Article CAS Google Scholar - Selin, L. K. et al. Attrition of T cell memory: selective loss of LCMV epitope-specific memory CD8+ T cells following infections with heterologous viruses. Immunity 11, 733–742 (1999).
Article CAS Google Scholar - Greten, T. F. et al. Direct visualization of antigen-specific T cells: HTLV-1 Tax11-19- specific CD8+ T cells are activated in peripheral blood and accumulate in cerebrospinal fluid from HAM/TSP patients. Proc. Natl Acad. Sci. USA 95, 7568–7573 (1998).
Article CAS Google Scholar - Welsh, R. M. in Encyclopedia of Virology (eds Granoff, A. & Webster, R. G.) 915–925 (Academic Press, New York, 1999).
Book Google Scholar - Welsh, R. M. in Effects of Microbes on the Immune System (eds Cunningham, M. W. & Fujinami, R. S.) 289–312 (Lippincott William & Wilkins, Philadephia, 2000).
Google Scholar - Marrie, T. J. & Saron, M. F. Seroprevalence of lymphocytic choriomeningitis virus in Nova Scotia. Am. J. Trop. Med. Hyg. 58, 47–49 (1998).
Article CAS Google Scholar - Whitton, J. L., Southern, P. J. & Oldstone, M. B. Analyses of the cytotoxic T lymphocyte responses to glycoprotein and nucleoprotein components of lymphocytic choriomeningitis virus. Virology 162, 321–327 (1988).
Article CAS Google Scholar - van der Most, R. G. et al. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology 240, 158–167 (1998).
Article CAS Google Scholar - Blattman, J. N., Sourdive, D. J., Murali-Krishna, K., Ahmed, R. & Altman, J. D. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J. Immunol. 165, 6081–6090 (2000).
Article CAS Google Scholar - Fenner, F., Henderson, D. A., Arital, I., Ladnyi, I. D. & Jezek, Z. Smallpox and its Eradication (World Health Organization, Geneva, 1988).
Google Scholar - Tufariello, J., Cho, S. & Horwitz, M. S. The adenovirus E3 14.7-kilodalton protein which inhibits cytolysis by tumor necrosis factor increases the virulence of vaccinia virus in a murine pneumonia model. J. Virol. 68, 453–462 (1994).
CAS PubMed PubMed Central Google Scholar - Selin, L. K., Lin, M. Y., Varga, S. M. & Welsh, R. M. in Cytotoxic Cells: Basic mechanisms and medical applications (eds Sitkovsky, M. V. & Henkart, P. A.) 327–361 (Lippincott Williams & Wilkins, Philadelphia, 2000).
Google Scholar - Butz, E. A. & Bevan, M. J. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8, 167–175 (1998).
Article CAS Google Scholar - Selin, L. K., Santolucito, P. A., Pinto, A. K., Szomolanyi-Tsuda, E. & Welsh, R. M. Innate Immunity to Viruses: Control of Vaccinia Virus Infection by γ/δ T Cells. J. Immunol. 166, 6784–6794 (2001).
Article CAS Google Scholar - Tay, C. H. et al. The role of LY49 NK cell subsets in the regulation of murine cytomegalovirus infections. J. Immunol. 162, 718–726 (1999).
CAS PubMed Google Scholar - Muller, U. et al. Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921 (1994).
Article CAS Google Scholar - Mahalingam, S., Foster, P. S., Lobigs, M., Farber, J. M. & Karupiah, G. Interferon-inducible chemokines and immunity to poxvirus infections. Immunol. Rev. 177, 127–133 (2000).
Article CAS Google Scholar - Skinner, P. J., Daniels, M. A., Schmidt, C. S., Jameson, S. C. & Haase, A. T. Cutting edge: In situ tetramer staining of antigen-specific T cells in tissues. J. Immunol. 165, 613–617 (2000).
Article CAS Google Scholar - Haanen, J. B. et al. In situ detection of virus- and tumor-specific T-cell immunity. Nature Med. 6, 1056–1060 (2000).
Article CAS Google Scholar - Gairin, J. E., Mazarguil, H., Hudrisier, D. & Oldstone, M. B. Optimal lymphocytic choriomeningitis virus sequences restricted by H- 2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J. Virol. 69, 2297–2305 (1995).
CAS PubMed PubMed Central Google Scholar - Schlesinger, C., Meyer, C. A., Veeraraghavan, S. & Koss, M. N. Constrictive (obliterative) bronchiolitis: diagnosis, etiology, and a critical review of the literature. Ann. Diag. Pathol. 2, 321–334 (1998).
Article CAS Google Scholar - Ehl, S., Hombach, J., Aichele, P., Hengartner, H. & Zinkernagel, R. M. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J. Exp. Med. 185, 1241–1251 (1997).
Article CAS Google Scholar - Zarozinski, C. C. & Welsh, R. M. Minimal bystander activation of CD8+ T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 185, 1629–1639 (1997).
Article CAS Google Scholar - Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Article CAS Google Scholar - Champagne, P. et al. Skewed maturation of memory HIV-specific CD8+ T lymphocytes. Nature 410, 106–111 (2001).
Article CAS Google Scholar - Rickinson, A. B. & Kieff, E. in Virology (eds Fields, B. N. et al.) 2397–2446 (Lippincott-Raven Publishers, Philadephia, 1996).
Google Scholar - Weinstein, L. & Meade, R. H. Respiratory manifestations of chickenpox. Arch. Intern. Med. 98, 91–99 (1956).
Article CAS Google Scholar - Simonsen, L. et al. Pandemic versus epidemic influenza mortality: a pattern of changing age distribution. J. Infect. Dis. 178, 53–60 (1998).
Article CAS Google Scholar - Amara, R. R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
Article CAS Google Scholar - Seth, A. et al. Recombinant modified vaccinia virus Ankara-simian immunodeficiency virus gag pol elicits cytotoxic T lymphocytes in rhesus monkeys detected by a major histocompatibility complex class I/peptide tetramer. Proc. Natl Acad. Sci. USA 95, 10112–10116 (1998).
Article CAS Google Scholar - Shirakawa, T., Enomoto, T., Shimazu, S. & Hopkin, J. M. The inverse association between tuberculin responses and atopic disorder. Science 275, 77–79 (1997).
Article CAS Google Scholar - Erb, K. J., Holloway, J. W., Sobeck, A., Moll, H. & Le Gros, G. Infection of mice with Mycobacterium bovis-Bacillus Calmette-Guerin (BCG) suppresses allergen-induced airway eosinophilia. J. Exp. Med. 187, 561–569 (1998).
Article CAS Google Scholar - Martinez, F. D. et al. Asthma and wheezing in the first six years of life. N. Engl. J. Med. 332, 133–138 (1995).
Article CAS Google Scholar - Walzl, G., Tafuro, S., Moss, P., Openshaw, P. J. & Hussell, T. Influenza virus lung infection protects from respiratory syncytial virus-induced immunopathology. J. Exp. Med. 192, 1317–1326 (2000).
Article CAS Google Scholar - Mathew, A. et al. Dominant recognition by human CD8+ cytotoxic T lymphocytes of dengue virus nonstructural proteins NS3 and NS1.2a. J. Clin. Invest. 98, 1684–1691 (1996).
Article CAS Google Scholar - Khalil, N. & Greenberg, A. H. Natural killer cell regulation of murine embryonic pulmonary fibroblast survival in vivo. Cell Immunol. 120, 439–449 (1989).
Article CAS Google Scholar - Mylin, L. M. et al. Quantitation of CD8(+) T-lymphocyte responses to multiple epitopes from simian virus 40 (SV40) large T antigen in C57BL/6 mice immunized with SV40, SV40 T-antigen-transformed cells, or vaccinia virus recombinants expressing full-length T antigen or epitope minigenes. J. Virol. 74, 6922–6934 (2000).
Article CAS Google Scholar
Acknowledgements
We thank S. S. Tevethia for assistance with the MHC tetramer protocols; D. M. Pardoll, J. P. Schneck and K. A. Kraemer for providing reagents, protocols and help in making IgG1 MHC dimers; and Y. Liu for technical assistance. Supported by National Institutes of Health research grants AR-35506 (to R. M. W.), AI-46578 (to L. K. S.) and Center Grant DK32520 (to I. J.). The contents of this publication are solely the responsibility of the authors and do not represent the official view of the National Institute of Health.
Author information
Authors and Affiliations
- Department of Pathology, University of Massachusetts Medical School, Worcester, 01655, MA, USA
Hong D. Chen, Armando E. Fraire, Isabelle Joris, Michael A. Brehm, Raymond M. Welsh & Liisa K. Selin
Authors
- Hong D. Chen
You can also search for this author inPubMed Google Scholar - Armando E. Fraire
You can also search for this author inPubMed Google Scholar - Isabelle Joris
You can also search for this author inPubMed Google Scholar - Michael A. Brehm
You can also search for this author inPubMed Google Scholar - Raymond M. Welsh
You can also search for this author inPubMed Google Scholar - Liisa K. Selin
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toLiisa K. Selin.
Rights and permissions
About this article
Cite this article
Chen, H., Fraire, A., Joris, I. et al. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung.Nat Immunol 2, 1067–1076 (2001). https://doi.org/10.1038/ni727
- Received: 16 July 2001
- Accepted: 18 September 2001
- Published: 22 October 2001
- Issue Date: 01 November 2001
- DOI: https://doi.org/10.1038/ni727