Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections (original) (raw)
Main
The mechanisms involved in inducing and maintaining hypergammaglobulinemia and autoantibody production associated with infection are largely unknown1,2,3,4,5,6,7. In B cell–tropic murine gammaherpesvirus, it has been shown that isotype switching during virus-induced hypergammaglobulinemia requires help from CD4+ T cells, which recognize peptide presented on major histocompatibility complex (MHC) class II molecules by virally infected B cells1. For viruses that do not usually infect B cells, such as lactate dehydrogenase–elevating virus (LDV) or lymphocytic choriomeningitis virus (LCMV) in mice and HIV or hepatitis viruses2,3,4 in humans, however, the mechanism of hypergammaglobulinemia induction is unclear. Interleukin 6 (IL-6) has been implicated in several syndromes associated with hypergammaglobulinemia5,6.
Hypergammaglobulinemia and autoantibody responses occur after murine infection with LCMV7,8,9, a natural noncytopathic segmented RNA virus of mice. Distinct LCMV isolates (WE, Armstrong, Docile, Clone 13) differ with respect to their tendency to establish a persistent infection in immunocompetent adult mice10,11. The LCMV genome comprises a small (S) and a large (L) ambisense RNA encoding the glycoprotein and nucleoprotein (S-RNA) and the polymerase (L-RNA), respectively12. Studies with reassortant viruses combining the S- and the L-RNA from persistence-prone and persistence-resistant strains have detected a dominance of the L-RNA12.
Here we have studied hypergammaglobulinemia and autoantibody responses after murine infection with LCMV7,8,9 to analyze the mechanisms underlying LCMV-induced hypergammaglobulinemia in mice.
Results
Influence of virus and host parameters
Infection of C57BL/6 mice with 2 × 106 plaque-forming units (PFU) of LCMV strain WE (LCMV-WE) resulted in a five- to tenfold increase in the serum concentrations of total immunoglobulin as compared with naive mice. The response peaked by day 12 (Fig. 1a). The hypergammaglobulinemia was polyclonal, as assessed by isoelectric focusing (data not shown). The increase in serum immunoglobulin concentration varied little between individual mice (naive mice, 1.31 ± 0.24 g/l (n = 6); day 12, mean 9.35 ± 1.13 g/l (n = 7); day 20, mean 6.65 ± 0.95 g/l (n = 9)). Thus, sera were pooled from three or four mice in subsequent experiments.
Figure 1: Correlation between viral replication and hypergammaglobulinemia.
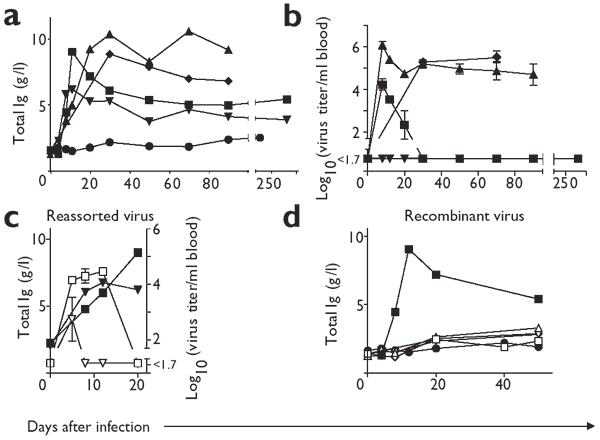
(a) C57BL/6 mice were infected with 2 × 106 PFU of LCMV strain Armstrong (▾), Docile (▴), Clone 13 (♦) or WE (▪). Uninfected C57BL/6 mice (●) were used as a control. The concentration of total immunoglobulin was measured at the indicated time points. (b) Blood viral titer was were measured at the indicated time points after C57BL/6 mice were infected with 2 × 106 PFU of LCMV strain Armstrong (▾), Docile (▴), Clone 13 (♦) or WE (▪). (c) C57BL/6 mice were infected with 2 × 106 PFU of a reassortant virus combining WE S-RNA and Armstrong L-RNA (▾) or Armstrong S-RNA and WE L-RNA (□). Filled symbols (▾, ▪) show the total immunoglobulin concentrations and open symbols (▿, □) represent LCMV titers in blood. (d) C57BL/6 mice were infected with 2 × 106 PFU of C57BL/6 mice were infected with 2 × 106 PFU of VSV-IND (□), Vaccinia-WR (▵),Vacc-G2 (▿) or Vacc-YN4 (⋄). For comparison, data for LCM-WE (▪, 2 × 106 PFU) are shown. Naive C57BL/6 mice (●) were used as a control. Immunoglobulin concentration was measured at the indicated time points. Three or four mice were used for each experiment; results are the mean of pooled sera (a,c,d) or the mean ± s.d. (b).
We next evaluated dependence of the extent of hypergammaglobulinemia on LCMV strains and doses. Infection of C57BL/6 mice with 2 × 106 PFU of LCMV strains Docile, Clone 13 and Armstrong induced a five- to tenfold increase in immunoglobulin within 12–20 days. Hypergammaglobulinemia remained high after infection with Docile or Clone 13 strains for 80–90 days, in parallel with high persistent viral loads (Fig. 1a,b).
Consistent with previous work11, we found that LCMV persistence exhausted virus-specific CD8+ cytotoxic T lymphocytes (data not shown). Infecting C57BL/6 mice with a low dose of 200 PFU of strains WE, Docile and Armstrong caused a delayed and limited peak in hypergammaglobulinemia at about day 20 (data not shown), despite the fact that infection with 200 PFU of WE, Armstrong, Clone 13 and Docile was rapidly cleared to below detection limits from blood and organs (data not shown).
The increase in immunoglobulin fraction was predominantly caused by large amounts of total IgG, which had increased by a factor of 5–10 on day 12, whereas total IgM had changed only a little (from 0.186 ± 0.122 g/l to 0.203 ± 0.063 g/l on day 12 after infection; data not shown). The serum concentration of total IgA increased two- to fourfold (data not shown).
We further confirmed the correlation between viral load and hypergammaglobulinemia in the following experiment. Reassortant viruses combining the WE strain S-RNA and the Armstrong strain L-RNA both induced lower virus titers in blood and reduced hypergammaglobulinemia as compared with reassortant viruses combining the Armstrong strain S-RNA and the WE strain L-RNA on day 20 after infection (Fig. 1c).
We assessed genetic predisposition to develop hypergammaglobulinemia after infection with 2 × 106 PFU of LCMV-WE in BALB/c, 129/Sv, DBA/2 and C3H eB/FeJ mice. All of these mouse strains showed increases in immunoglobulin concentration independent of their genetic host background (Table 1). By contrast, infection of C57BL/6 mice with vesicular stomatitis virus (VSV), wild-type vaccinia virus (Vacc) and a recombinant vaccinia virus expressing the LCMV nucleoprotein (Vacc-YN4) or glycoprotein (Vacc-G2) did not show an increase in immunoglobulin concentration (Fig. 1d). Thus, non- or poorly replicating viruses, such as those tested, do not induce hypergammaglobulinemia, whereas widely replicating viruses with a tendency to persist, such as LCMV or LDV, do.
Table 1 Hypergammaglobulinemia in inbred and mutant mouse strains
Polyclonality of LCMV-induced hypergammaglobulinemia
Analysis of the antibody specificities showed that the specific IgG response to anti–LCMV nucleoprotein did not correlate with an increase in concentration of total immunoglobulin after infection with a low or high dose of LCMV (data not shown). The anti–LMCV nucleoprotein response peaked on day 8–12 and remained stable thereafter up to day 250, whereas the serum concentration of total immunoglobulins peaked around day 12 and declined thereafter (Fig. 1a). We confirmed the polyclonality of the increased immunoglobulins by isoelectric focusing (data not shown).
To assess B cell specificities, we examined the percentages of LCMV-specific B cells in the spleen 12 days after intravenous (i.v.) infection of C57BL/6 mice with 200 PFU or 2 × 106 PFU of LCMV-WE, or 2 × 106 PFU of Vacc-YN4, by using an LCMV nucleoprotein–specific enzyme-linked immunospot (ELISPOT) assay (Table 2). On day 12 after infection with LCMV-WE, the total number of B cells producing IgG had increased 50- to 100-fold as compared with naive mice, and 10- to 20-fold as compared with mice infected with Vacc-YN4. By contrast, after infection with 2 × 106 PFU of LCMV-WE, only 1–3% of the total IgG-producing cells were specific for LCMV nucleoprotein, the most abundant LCMV protein (Table 2). Thus, more than 90% of the IgG-producing cells seemed to be nonspecific for LCMV.
Table 2 LCMV-specific and -nonspecific B cell responses
Influence of cytokines and B cell receptors
Interferons, IL-6 and other cytokines have been reported to have a stimulatory role in B cell responses. We therefore evaluated the influence of cytokines on LCMV-induced hypergammaglobulinemia after infecting various cytokine-deficient mouse strains with 2 × 106 PFU of LCMV-WE (Table 1). The absence of T helper type 1 (TH1)-specific interferon-γ functions13, TH2-specific IL-4 (ref. 14), the interferon-α/β receptor15 or IL-12 did not influence the immunoglobulin increase in mice infected with LCMV (Table 1). The initial peak of serum immunoglobulins on days 12–20 after infection was absent in IL-6−/− mice16, but at later time points the immunoglobulin concentrations were comparable to those in wild-type C57BL/6 mice infected with LCMV-WE (data not shown).
To evaluate the role of the B cell receptor (BCR) specificity and its co-receptors in possible uptake of viral antigen, we infected CD19−/− mice17, CR2 (CD21)−/− mice18, IgM−/− mice (which lack IgM but have IgD on the cell surface and can generate IgG)19 and C3−/− mice (which lack the ligand for the CD21 receptor)20. These mouse strains and FcγRI-III−/− mice showed hypergammaglobulinemia on infection with 2 × 106 PFU of LCMV-WE (Table 1). Thus, we found little evidence for cytokine involvement; in addition, neither specific immunoglobulin nor any of the other BCRs tested seemed to have a prominent role in LCMV-induced hypergammaglobulinemia.
Induction of virus-nonspecific B cells
We assumed that much of the increase in immunoglobulin should reflect IgM switching to IgG in preimmune B cells with receptors that were nonspecific for LCMV antigens. We assessed this in quasimonoclonal (QM) mice21, in which 80% of the B cells are specific for the hapten (4-hydroxy-3-nitrophenyl)acetate (hereafter abbreviated as nitrophenyl). The possibility that LCMV might bind to and infect QM B cells through the nitrophenyl-specific receptor was unlikely because QM serum did not bind to LCMV glycoprotein or nucleoprotein in an enzyme-linked immunosorbent assay (ELISA; data not shown). After infection with LCMV-WE, the anti-nitrophenyl IgG concentration increased by 64-fold (Fig. 2a). Note that the amount of total IgG needed for the same absorption value in an anti-nitrophenyl ELISA was 4–5-fold higher in noninfected than in infected QM mice, indicating an absolute increase of anti-nitrophenyl IgG (Fig. 2a). Nitrophenyl-specific IgG was also detectable in LCMV-WE–infected but not uninfected C57BL/6 mice (Fig. 2a). Natural concentrations of anti-nitrophenyl IgM were high in QM mice and did not change substantially after infection with LCMV-WE (Fig. 2b).
Figure 2: Nonspecific B cells are activated and switch to IgG after infection with LCMV.
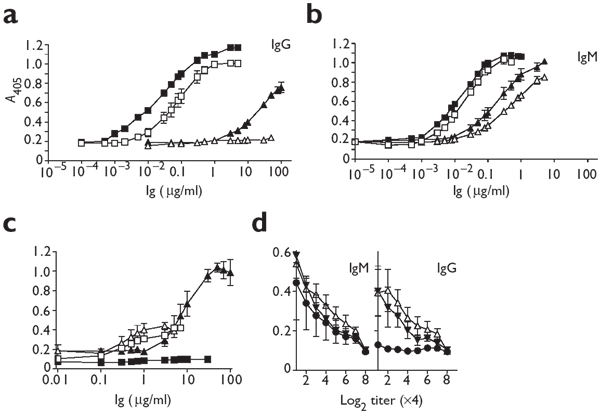
(a,b) Serum from either naive C57BL/6 (▵) or QM (□) mice, or from C57BL/6 (▴) or QM (▪) mice on day 12 after infection with 2 × 106 PFU of LCMV-WE was analyzed for nitrophenyl-specific IgG (a) or IgM (b). (c) Serum from naive C57BL/6 mice (□, ▪) or C57BL/6 mice infected with 2 × 106 PFU of LCMV-WE (▵, ▴) was analyzed by ELISA for titers of IgM (▵, □) and IgG (▴, ▪) specific for purified VSV-IND. (d) Serum from naive C57BL/6 mice (●) or C57BL/6 mice infected with 2 × 106 PFU of LCMV-WE (▾) or LDV (▵) was analyzed for IgG or IgM specific for influenza HA. Results are the mean ± s.d. from the analysis of three mice per group.
To test further the specificity of polyclonal IgG responses, we measured the induction of other non-LCMV and natural antibody specificities in C57BL/6 mice infected with LCMV. IgG specific for VSV antigens was detectable by ELSIA 12 days after infection with LCMV-WE (Fig. 2c). Concentrations of anti-VSV IgM increased only marginally (Fig. 2c). We also tested sera from mice infected with LCMV-WE (on day 12) and LDV (on day 30) by ELISA for specificities to influenza hemagglutinin (HA). Both viruses induced an increase in titers of anti-HA IgG, whereas those of anti-HA IgM remained comparable (Fig. 2d).
Because release of lipopolysaccharide (LPS) from commensal intestinal bacteria as a result of viral enteropathy might underlie the polyclonal B cell stimulation seen in hypergammaglobulinemia, we examined the effect in germ-free (gnotobiotic) mice. We found that the hypergammaglobulinemia and titers of IgG specific for the tested antigens were comparable to those in specific pathogen–free (SPF) C57BL/6 mice infected with LCMV-WE (Table 1). The absence of LPS-induced effects was confirmed by comparable increases in serum immunoglobulin in LPS-responder (C3HeB/FeJ) and nonresponder (C3H/HeJ) mouse strains8 (Table 1).
Induction of autoantibodies
The increase in IgG specific for nonviral antigens after infection with LCMV prompted us to evaluate virus-induced autoantibody responses. Transgenic MONITOR mice22 expressing a soluble form of the glycoprotein from VSV serotype Indiana (VSV-IND) did not develop VSV autoantibodies when immunized with ultraviolet-inactivated VSV-IND (Supplementary Fig. 1 online). By contrast, the same mice mounted an IgG response to the VSV-IND glycoprotein when infected with LCMV. This IgG response was enhanced when ultraviolet-inactivated VSV-IND plus LCMV-WE were co-injected (Supplementary Fig. 1 online).
This observation suggested that autoreactive B cell responses of high affinity22 were induced through linked LCMV-specific TH cells. In addition, the numbers of IgG autoantibodies to phosphorylcholine23 were increased after infecting BALB/c mice with LCMV-WE (Supplementary Fig. 1 online). Similarly, we assessed the production of autoantibodies to autoantigens implicated in human diseases. In sera of LCMV-infected mice (Fig. 3), we found increased titers of IgGs to thyroglobulin (>1:270), double-stranded DNA (dsDNA; between 1:32 and 1:64), single-stranded DNA (ssDNA; >1:90) and insulin (>1:145).
Figure 3: Induction of autoantibodies after LCMV infection.
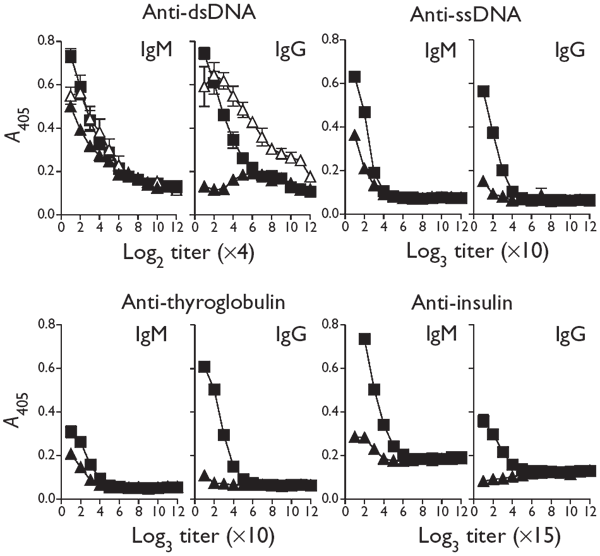
IgM and IgG specific for autoantigen were measured in naive mice (▴), on day 12 after infection with LCMV (▪) and on day 30 after infection with LDV (▵). The indicated predilutions were used. Results are the mean ± s.d. of antigen-specific IgM or IgG titers from three mice per group.
Role of virus-specific CD4+ T cells
We found that the induction of hypergammaglobulinemia by LCMV was dependent on CD4+ T cells. T cell receptor (TCR)-deficient mice lacking the β and δ TCR chains (TCRβδ−/−) and athymic T cell–deficient nude (nu/nu) mice did not develop hypergammaglobulinemia after infection with LCMV-WE (Fig. 4a). Mice lacking surface CD40L, which is crucial for immunoglobulin isotype switching and germinal center formation, also did not develop hypergammaglobulinemia after infection with LCMV-WE (Fig. 4a).
Figure 4: Specific CD4+ T cells are required to induce hypergammaglobulinemia.
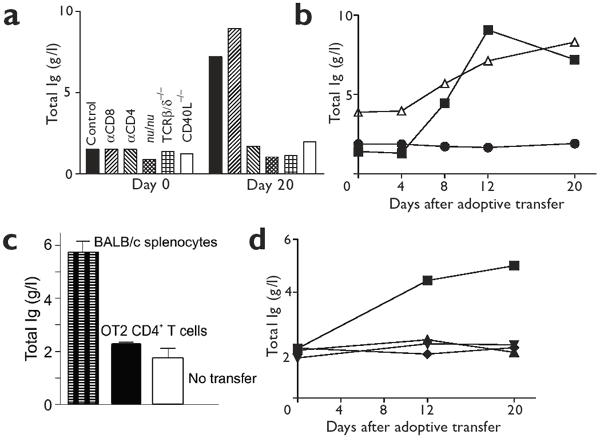
(a) Untreated C57BL/6 mice or C57BL/6 mice treated with depleting anti-CD8 or anti-CD4, C57BL/6 nu/nu mice, TCRβδ−/− mice or CD40L−/− mice were infected with 2 × 106 PFU of LCMV-WE, and serum immunoglobulin was measured. Serum was pooled from naive mice (day 0) and from mice 20 days after infection (three mice per group). (b) Naive Smarta1 CD4+ TCR transgenic T cells (1 × 107; specific for the LCMV GP61 peptide) were adoptively transferred into LCMV-WE carrier mice (▵), and the total serum immunoglobulin was measured in pooled sera (three mice per group). Naive C57BL/6 mice that had received 1 × 107 naive Smarta1 CD4+ TCR transgenic T cells (●) were used as controls. For comparison, data for LCMV-WE (▪, 2 × 106 PFU) are shown. (c) BALB/c nu/nu mice (no transfer), BALB/c nu/nu mice adoptively transferred with 1 × 107 BALB/c wild-type splenocytes on day −1, or BALB/c nu/nu mice adoptively transferred with 1 × 107 BALB/c OT2 × RAG1−/− cells on day −1 were infected with 2 × 106 PFU of LCMV-WE, and the total serum immunoglobulin was measured on day 12. Results are the mean ± s.d. from the analysis of four mice per group. (d) Naive (♦, ▴) or _in vitro_–activated (cultured for 4 d with GP61 peptide; ▪, ▾) Smarta1 CD4+ TCR transgenic T cells (1 × 107) were adoptively transferred into DEE mice (▪, ▴), which express the LMCV glycoprotein, or C57BL/6 mice (♦, ▾) and total immunoglobulin concentrations were measured at the indicated time points in sera pooled from three mice per group.
To test whether virus-specific CD4+ cells were required, we transferred monoclonal transgenic CD4+ T cells specific for the major TH epitope of the LCMV glycoprotein (Smarta1)24 into chronically LCMV-WE–infected mice (LCMV carrier mice). This transfer induced hypergammaglobulinemia with similar kinetics and magnitude to those seen in C57BL/6 mice infected with LCMV (Fig. 4b). By contrast, ovalbumin-specific CD4+ T cells from BALB/c OT2 transgenic × RAG1−/− mice transferred into T cell–deficient BALB/c nu/nu mice did not induce hypergammaglobulinemia after infection with LCMV-WE, whereas the transfer of control naive BALB/c splenocytes did induce an increase in immunoglobulin concentrations (Fig. 4c).
To examine whether under special conditions hypergammaglobulinemia is inducible by nonreplicating antigen, we adoptively transferred Smarta1 splenocytes into transgenic DEE mice, which ubiquitously express the LCMV glycoprotein25. A threefold increase in immunoglobulin concentration was seen in DEE mice only when Smarta1 cells were preactivated in vitro before transfer. Neither _in vitro_–preactivated nor naive Smarta1 cells induced an increase in immunoglobulin concentration in uninfected C57BL/6 control mice (Fig. 4d).
Specific cognate T cell–B cell cooperation
IgG responses generally require cognate CD4+ T cell–B cell cooperation through specific peptides carried on MHC class II molecules of B cells. It was therefore essential to evaluate whether B cells presented viral peptides through MHC class II molecules regardless of BCR specificity. To address this issue, we generated mixed chimeras by transferring bone marrow cells from μMT (1 × 107 cells) and MHC class II−/− (1 × 107 cells) donors together into irradiated (950 rad) C57BL/6 mice. The μMT donors provided MHC class II+ cells of hematopoietic origin except for B cells; the Ig+ B cells were all MHC class II−/−.
These chimeras generated neutralizing antibodies to VSV26, which verified unimpaired B cell function (data not shown). We used irradiated C57BL/6 mice reconstituted with C57BL/6 bone marrow cells as controls. After infection with LCMV-Docile, the control chimeras, but not the experimental mixed chimeras, induced hypergammaglobulinemia within 12 days (Fig. 5a). Thus, expression of MHC class II molecules on B cells is essential for the increased immunoglobulin response.
Figure 5: Virus-nonspecific B cells present LCMV-derived peptides on MHC class II.
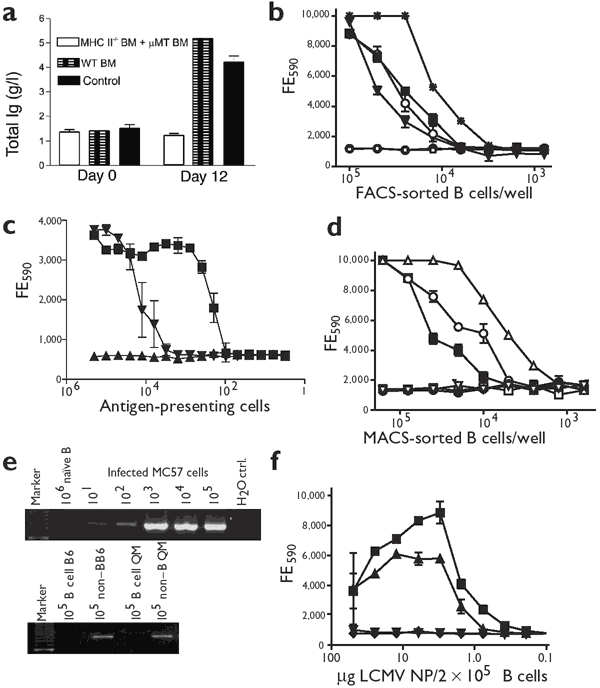
(a) Lethally irradiated C57BL/6 mice received bone marrow cells from either MHC class II−/− and μMT mice or from wild-type (WT) C57BL/6. Four mice per group were infected with 2 × 106 PFU of LCMV-Docile. Untreated C57BL/6 mice were used as controls. Serum samples were collected at the indicated time points and total immunoglobulin was measured. (b) Total B cells from naive C57BL/6 mice (□), C57BL/6 mice on day 9 after infection with 2 × 106 PFU of LCMV-WE (▪), or nitrophenyl-specific B cells from naive (●) or infected (○) QM mice were sorted by FACS. Total splenocytes (sorted on living cells; *) from C57BL/6 mice on day 5 after infection with 2 × 106 PFU of LCMV-WE and B cells from C57BL/6 LCMV-WE carrier mice (▾) were used as positive controls. Diluted FACS-sorted cells were incubated with the VE8 CD4+ T cell hybridoma (specific for LCMV nucleoprotein), and IL-2 secretion in the supernatant was measured by assessing the viability of IL-2–sensitive CTLL cells with Alamar blue. (c) DCs labeled with NP309 peptide (▪) were titrated into naive MACS-sorted B cells and IL-2 secretion by the VE8 CD4+ T cell hybridoma was measured as in b. Splenocytes 5 d after infection with LCMV (▾) and naive splenocytes (▴) were used as positive and negative controls, respectively. (d) Total B cells from naive C57BL/6 mice (□), C57BL/6 mice on day 9 after infection with 2 × 106 PFU of LCMV-WE (▪), or nitrophenyl-specific B cells from QM mice (●, naive; ○, infected) were sorted by MACS. Naive B cells were incubated with GP61 (▿) or NP309 (▵) peptide. Diluted MACS-sorted cells were incubated with the VE8 CD4+ T cell hybridoma and analyzed as in b. (e) RT-PCR for genomic RNA from LCMV-WE. LCMV-infected MC57 cells (48 h after infection) were diluted into uninfected B cells; naive B cells and H2O were used as negative controls (top). RT-PCR on FACS-sorted B cells or on the non–B cell fraction of day 9 immune C57BL/6 or QM mice (bottom) are shown. (f) μMT-T11 (▪) and C57BL/6 (▴) B cells were incubated with various concentrations of recombinant LCMV nucleoprotein (NP). IL-2 secretion by the VE8 CD4+ T cell hybridoma was assessed as in b. LCMV nucleoprotein (▾) alone and VE8 cells (♦) alone were used as controls.
Virus-nonspecific B cells present viral peptides
To examine whether B cells specific for antigens other than LCMV can present MHC class II–restricted LCMV peptides, we purified nitrophenyl-specific B cells by fluorescence-activated cell sorting (FACS) from QM mice that had been infected 9 days earlier with LCMV-WE. We used total B cells from infected and from uninfected C57BL/6 mice as controls. After infection with LCMV-WE, C57BL/6 and QM nitrophenyl-specific B cells stimulated the LCMV nucleoprotein–specific TH hybridoma VE8 (which recognizes the nucleoprotein-derived NP309 peptide on I-Ab) to secrete IL-2, whereas B cells from naive mice did not (Fig. 5b).
Because the purity of the FACS-sorted B cells was around 99%, we could not exclude the possibility of minor contamination with dendritic cells (DCs). We therefore repeated the above experiment with DCs that had been incubated with NP309 peptide, which we titrated into naive magnetically sorted (MACS) B cells. Peptide-labeled DCs were 50-fold more effective on a per cell basis than were FACS-sorted B cells in stimulating the hybridoma27 (Fig. 5c).
We evaluated the extent of LCMV peptide presentation by anti-nitrophenyl–specific QM or C57BL/6 control B cells from hypergammaglobulinemia-developing mice infected with LCMV-WE. These cells were compared with naive C57BL/6-B cells labeled in vitro with LCMV NP309 peptide for their respective capacity to induce VE8 CD4+ T cell hybridoma cells to secrete of IL-2. For numeric comparison, about 10% of the MACS-sorted B cells seemed to present viral peptide (Fig. 5d). Because the concentration of peptide used for pulsing naive B cells influences the dose-response curve, however, equivalence could not be established on a per cell basis.
We next examined whether LCMV antigen or peptides were taken up from the outside or were synthesized by infected B cells themselves. We used a nested semi-quantitative approach based on polymerase chain reaction with reverse transcriptase (RT-PCR) that could detect ten LCMV-infected MC57 fibroblasts (48 h after infection) in a million cells (Fig. 5e). No PCR signal was detected in 1 × 105 FACS-sorted B cells from LCMV-infected C57BL/6 or QM mice (Fig. 5e). By contrast, viral RNA was present in 105 cells from the non–B cell fraction of splenocytes of the same mice (Fig. 5e). Thus, pathways other than active infection must be involved in the presentation of LCMV NP309 peptide by B cells.
We assessed whether nonreplicating viral antigen could be taken up independently of the specificity of the BCR and presentation on MHC class II molecules. For this approach, we used a transgenic mouse carrying a gene encoding a neutralizing anti-VSV IgM backcrossed to μMT mice (μMT-T11)28. The μMT-T11 mouse can make only VSV-neutralizing IgM antibodies28. We sorted μMT-T11 B cells and C57BL/6 B cells by magnetic separation and incubated them for 24 h with different concentrations of the recombinant LCMV nucleoprotein. LCMV nucleoprotein could be taken up by the μMT-T11 B cells nonspecifically down to a concentration of 2 μg/ml and was presented to the NP309-specific T cell hybridoma VE8, which then secreted IL-2 (Fig. 5f). Similarly treated C57BL/6 B cells presented the NP309 epitope efficiently, in contrast to untreated control B cells, which did not induce a response (Fig. 5f). Thus, LCMV nucleoprotein can be taken up and presented by B cells irrespective of their BCR specificity.
Discussion
We have characterized some key parameters of hypergammaglobulinemia provoked by systemic and persistent viral infection. Viral replication and persistence were correlated with an increased concentration of immunoglobulin, mostly of the IgG isotype. The highest gamma-globulin concentrations were induced by the LMCV strains Docile and Clone 13, which resulted in a carrier state; by contrast, the Armstrong strain, which is usually rapidly cleared, caused only a limited increase in immunoglobulin concentrations in normal mice.
LCMV-induced hypergammaglobulinemia was polyclonal and only a minor fraction of the activated B cells produced virus-specific antibodies. Our results suggest that the diverse specificities of IgG provoked by LCMV infection result from immunoglobulin class switching from IgM produced in the preimmune (natural) repertoire. This was shown in the QM mouse21, by the classical 'natural' anti-phosphorylcholine specificity found in the wild-type BALB/c strain23 and by the switching of natural IgM specific for influenza or VSV to IgG.
Given the diversity of the switching process in hypergammaglobulinemia, potentially harmful autoantibodies or immune complexes may be produced. Several studies have documented enhanced autoantibody-mediated autoimmunity after infection with LCMV7,8,9. Antibody-mediated immunopathology is also found in human autoimmune and chronic viral diseases. Deposits of immune complexes in kidney glomeruli are found in hepatitis B virus–associated glomerulonephritis29, and immune complex–mediated cryoglobulinemia is associated with chronic hepatitis C virus infections30. HIV infections often show a polyclonal serum hypergammaglobulinemia, and antibodies to platelet antigens can lead to HIV-related immune thrombocytopenia31. In humans, hypergammaglobulinemia is often associated with inflammatory disorders in which replicating antigens are not characterized, such as systemic lupus erythematosus or rheumatoid arthritis.
Using the transgenic DEE mouse (which expresses LCMV glycoprotein), we have shown here that nonreplicating and therefore limiting antigen can be sufficient to induce hypergammaglobulinemia when specific activated T cell help against an autoantigen is present. The observed hypergammaglobulinemia required virus-specific T cell help through interactions between the TCR and MHC class II molecules, in combination with CD40–CD40L binding. Similar cognate CD4+ T cell–B cell interactions have been shown in a study where T cell help was induced through the delivery of a ubiquitous maleylated immunoglobulin self-protein by scavenger receptors, resulting in a transient hypergammaglobulinemia32.
Dendritic cells ameliorate antigen transfer to B cells and initiate class switching in a T cell–dependent response33; they also influence the differentiation into and survival of plasmablasts27. We cannot exclude the possibility that DCs are involved in the LCMV-induced hypergammaglobulinemia responses in a three-way type of DC-improved interaction between B and T cells33. Our experiments show, however, that B cells can present antigen to T cells on MHC class II molecules irrespective of the BCR specificity.
The mechanism by which B cells present foreign MHC class II peptide remains to be determined. Several mechanisms might explain how B cells could present viral antigens: for example, infection of B cells, BCR-independent uptake of viral antigen through complement or Fc receptor, or uptake of viral proteins through antigen-nonspecific pinocytosis. Although we did not detect LCMV genomic RNA in 105 B cells of infected mice using a PCR-based assay sufficiently sensitive to detect fewer than ten infected cells, we cannot completely exclude the possibility that there was very low-level infection in those cells.
The uptake of viral antigen through complement or Fc receptor did not seem to be essential for LCMV-provoked hypergammaglobulinemia, because hypergammaglobulinemia was still induced in CR2−/−, C3−/− and FcγR I-III−/− mice. By contrast, defects in several complement factors34 and in the reticuloendothelial system35 are associated with human autoimmune diseases and hypergammaglobulinemia-associated syndromes. This difference may be due to the rather short-term nature of our experiments as compared with the course of human autoimmune diseases.
Our experiments suggest that a nonspecific pinocytosis mechanism may be involved. Under conditions in which sufficient antigen concentrations are generated during an acute or persistent infection, B cells may be permitted to undergo MHC class II loading of peptide independent of BCR specificities36. It is obvious that high amounts of antigen are needed to induce hypergammaglobulinemia and it is noteworthy that HIV, LDV and LCMV all show a tropism for lymphoid organs and macrophages. Our data establish a pathway indicating how hypergammaglobulinemia and autoantibodies can be induced by LCMV. These findings may be generalized to other viral infections associated with hypergammaglobulinemia, autoantibodies and immune complex–linked disease in humans.
Methods
Mice.
Sex-matched C57BL/6, BALB/c, DBA/2, 129Sv, DEE, μMT, SmartaI-II TCR transgenic, MONITOR, TCR-βδ −/−, IL-4−/−, CD19−/−, IL-6−/−, IL-12−/−, CD40L−/−, IFN-αβ receptor−/−, IFN-γ receptor−/− and μMT-T11 mice were purchased from the Institute für Labortierkunde (Univ. of Zurich, Switzerland). We housed germ-free C57BL/6 mice in a sterile isolator at the Institute für Labortierkunde. C3H eB/FeJ and C3H HeJ mice were bought from Jackson Laboratories (Bar Harbor, ME). C57BL/6 nu/nu and BALB/c nu/nu were bought from Iffa Credo, Les Oncins, France. The generation of QM mouse has been described21. IgM−/− mice were a gift of F. Brombach and H. Mossmann (Max-Planck-Institute for Immunobiology, Freiburg, Germany); OT2 CD4+ transgenic × RAG1−/− mice were a gift of M. Kopf (Basler Institute for Immunology, Basel, Switzerland); MHC class II−/− were a gift of H. Blüthman (Basel, Switzerland); and CR2−/−, C3−/− and FcγRI-III−/− were a gift of M. Klein (Zürich, Switzerland). We bred and maintained mice under SPF conditions. All animal experiments were done in accordance with institutional and Swiss national regulations.
Virus.
LCMV strains WE, Armstrong and Docile were a gift of F. Lehmann-Grube (Hamburg, Germany) and C. Pfau (Troy, NY) and were propagated on L929, BHK and MDCK cells. Vaccinia virus strain WR and recombinant Vacc-YN4 and Vacc-G2 were injected i.v. at a dose of 2 × 106 PFU. VSV-IND was originally obtained from D. Kolakovsky (Univ. of Geneva, Switzerland). LCMV reassortant viruses were a gift of M.B.A. Oldstone (Scripps Research Institute, La Jolla, CA) and R. Ahmed (Emory Univ. School of Medicine, Atlanta, GA). For infection, 2 × 106 PFU of VSV-IND was injected i.v., or VSV-IND was inactivated by ultraviolet irradiation for 5 min and the equivalent of 5 × 107 PFU was injected i.v. LDV isolate P was obtained from P.G.W. Plagemann (Univ. of Minneapolis, Minnesota). LDV was injected i.v. and produced and assessed for titers as described37. In vivo depletion of CD4+ and CD8+ T cells was done with rat monoclonal antibodies against CD4 (YTS191.6) and against CD8 (YTS 169.4)38.
Bone marrow chimeras.
C57BL/6 mice were lethally irradiated with 950 rad and 1 × 107 bone marrow cells from MHC class II−/− and μMT mice were transferred i.v. to the mice. As a control, wild-type C57BL/6 bone marrow cells were transferred. After 90 d, we analyzed mice by FACS for the presence of CD4+ T cells, CD8+ T cells and the expression of I-Ab on total splenocytes and CD19+B220+ cells.
Nested RT-PCR.
RNA was extracted with TRIzol (Gibco-BRL, Basel, Switzerland) from 1 × 106 FACS-sorted B cells from naive C57BL/6 or QM spleens, splenocytes from C57BL/6 or QM mice 9 d after infection with 2 × 106 PFU of LCMV-WE, or splenocytes from LCMV C57BL/6 carrier mice. As a sensitivity control, MC57 fibroblasts infected with LCMV-WE (5 or 48 h after infection) were titrated into naive B cells. We did the RT reaction (5 μg total RNA) and nested PCR as described39. PCR products were visualized by ethidium bromide after 1% agarose gel electrophoresis.
FACS.
Single-cell spleen suspensions from naive C57BL/6 or QM mice, C57BL/6 or QM mice on day 9 after infection with LCMV-WE, or LCMV-WE C57BL/6 carrier mice were labeled with the phycoerythrin (PE)-labeled monoclonal antibody against mouse CD19 (1D3; Pharmingen, San Diego, CA). Cells from QM mice were also stained with fluorescein isothiocyanate (FITC)-labeled anti-idiotype reagent against anti-nitrophenyl21. We washed the cells and sorted them for CD19+ or CD19+ and anti-idiotype+ by FACS. The purity after sorting was more than 99%.
MACS.
We negatively sorted QM splenocytes for CD11b and CD11c (Miltenyi Biotec GmbH, Cologne, Germany) and then positively sorted them for anti-nitrophenyl by incubating them with biotinylated anti-idiotypic21 and Streptavidin MicroBeads (Miltenyi Biotec GmbH). Splenocytes from naive C57BL/6 mice or C57BL/6 on day 9 after infection with LCMV-WE (2 × 106 PFU) were incubated with B220 anti-mouse MicroBeads (Miltenyi Biotec GmbH) and sorted by MACS. The purity was assessed by FACS and found to be more than 93%.
ELISA.
ELISA assays for antigen-specific immunoglobulins, including those against nitrophenyl, VSV-IND, VSV glycoprotein, LCMV nucleoprotein, total IgG, IgG1, IgG2a, IgG2b, IgG3, IgM or IgA, were done as described40. For the anti-nitrophenyl assays, ELISA plates were coated with 1 μg/ml of nitrophenyl chicken γ-globulin (Biosearch Technologies, Novato, CA). The influenza HA-specific ELISA plates were coated with 10 μg/ml of trivalent influenza virosome chimeras containing A/Singapore/6/86-like (H1N1), A/Johannesburg/33/94-like (H3N2) and B/Beijing/184/93-like (Schweizerisches Serum & Impfinstiut, Bern, Switzerland). We obtained dsDNA and ssDNA from Sigma (Buchs, Switzerland) and used them at 0.01 mg/ml. Insulin (Sigma) and thyroglobulin (Sigma) were used at 0.25 mg/ml.
LCMV peptide presentation assays.
The T cell hybridomas VE8 and 5A1 have been described41. MACS- or FACS-sorted B cells or total splenocytes were irradiated (2,000 rad) and diluted 1:2 starting from 2 × 105 (MACS) or 1 × 105 (FACS) B cells incubated with 4 × 104 VE8 or 5A1 cells. After 24 h, 50 μl of supernatant was transferred and 1 × 104 IL2-sensitive CTLL cells were added. After 24 h, 9 μl of the viability dye Alamar blue (Biosource International, Camarillo, CA) was added and the fluorescence emission at 590 nm was measured after 6–8 h by a CytoFluor spectrophotometer (Millipore). Naive B cells were incubated for 1 h at 37 °C with 10−4 M of NP309 peptide and diluted thereafter as described above. We labeled bone marrow–derived DCs with 10 μg/ml of NP309 peptide and then titrated them into MACS-sorted naive B cells before the peptide presentation assay. We used naive splenocytes or splenocytes on day 5 after infection with LCMV as controls
LCMV nucleoprotein pinocytosis by LCMV-nonspecific B cells.
Recombinant LCMV nucleoprotein supernatants were treated with RNase and DNase for 15 min and serially diluted (1:2) starting from a concentration of 0.5 mg/ml. We added 2 × 105 MACS-sorted μMT-T11 or C57BL/6 B cells to each dilution and incubated them with 4 × 104 VE8 cells for 24 h. IL-2 production was measured as described for the LCMV peptide presentation assay.
Serum protein electrophoresis.
Electrophoresis of serum proteins was done by the Department of Clinical Chemistry, Univ. Hospital Zürich. The concentration (g/l) of total serum immunoglobulins was assessed by PAGE.
Isoelectric focusing.
We measured serum immunoglobulins by kinetic nephelometry (Beckmann Array 360 System, Milan, Italy) and diluted them in PBS plus 0.05% Tween. Isoelectric focusing was done with the CSF Analysis Kit (Amersham Pharmacia, Chalfont, UK). In brief, 20 ng of immunoglobulin was separated by PAGE. Immunofixation was done with a rabbit anti–human IgG (Dako, Glostrup, UK) or rabbit anti–mouse IgG (Zymed, San Francisco, CA) for 1 h and IgGs were visualized by silver staining.
ELISPOT assay.
We coated 24-well plates with rabbit anti–mouse IgM (Zymed; for total IgM), rabbit anti–mouse IgG (Zymed) or recombinant LCMV nucleoprotein. Splenocytes from uninfected C57/B6 mice or C57/B6 mice on day 12 or day 20 after infection with LCMV or Vacc-YN4 were serially diluted 1:5 and incubated for 5 h in serum-free medium. Rat anti–mouse IgM (Pharmingen) and goat anti–mouse IgG (Sigma) were used as secondary antibodies. The tertiary antibodies were alkaline phosphatase–conjugated donkey Fab fragment against rat IgM (Jackson Laboratories) and alkaline phosphatase–conjugated donkey antibodies against goat IgG (Jackson Laboratories). Spots were visualized with alkaline phosphatase buffer (Sigma) containing 1 mg/ml of 5-bromo-4-chloro-3-indolylphosphate (BCIP).
Note: Supplementary information is available on the Nature Immunology website.
References
- Sangster, M.Y. et al. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol. 164, 1820–1828 (2000).
Article CAS Google Scholar - Tirelli, U. et al. Persistent generalized lymphadenopathy: clinical characteristics of lymphadenopathy syndrome in intravenous drug abusers. AIDS Res. 2, 227–230 (1986).
Article CAS Google Scholar - Tsianos, E.V. et al. Oligoclonal immunoglobulin bands in serum in association with chronic viral hepatitis. Am. J. Gastroenterol. 85, 1005–1008 (1990).
CAS PubMed Google Scholar - Kawamoto, H. et al. Autoimmune responses as assessed by hypergammaglobulinemia and the presence of autoantibodies in patients with chronic hepatitis C. Acta. Med. Okayama 47, 305–310 (1993).
CAS PubMed Google Scholar - Hirano, T. et al. Human B-cell differentiation factor defined by an anti-peptide antibody and its possible role in autoantibody production. Proc. Natl. Acad. Sci. USA 84, 228–231 (1987).
Article CAS Google Scholar - Hirano, T. & Kishimoto, T. Interleukin 6 and plasma cell neoplasias. Prog. Growth Factor Res. 1, 133–142 (1989).
Article CAS Google Scholar - Oldstone, M.B. & Dixon, F.J. Pathogenesis of chronic disease associated with persistent lymphocytic choriomeningitis viral infection. I. Relationship of antibody production to disease in neonatally infected mice. J. Exp. Med. 129, 483–505 (1969).
Article CAS Google Scholar - Coutelier, J.P., Johnston, S.J., El Idrissi, M.E. & Pfau, C.J. Involvement of CD4+ cells in lymphocytic choriomeningitis virus-induced autoimmune anaemia and hypergammaglobulinaemia. J. Autoimmun. 7, 589–599 (1994).
Article CAS Google Scholar - Coutelier, J.P. et al. In vivo polyclonal B-lymphocyte activation elicited by murine viruses. J. Virol. 64, 5383–5388 (1990).
CAS PubMed PubMed Central Google Scholar - Thomsen, A.R., Johansen, J., Marker, O. & Christensen, J.P. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J. Immunol. 157, 3074–3080 (1996).
CAS PubMed Google Scholar - Moskophidis, D., Lechner, F., Pircher, H. & Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761 (1993).
Article CAS Google Scholar - Riviere, Y., Ahmed, R., Southern, P.J., Buchmeier, M.J. & Oldstone, M.B. Genetic mapping of lymphocytic choriomeningitis virus pathogenicity: virulence in guinea pigs is associated with the L RNA segment. J. Virol. 55, 704–709 (1985).
CAS PubMed PubMed Central Google Scholar - Huang, S. et al. Immune response in mice that lack the interferon-γ receptor. Science 259, 1742–1745 (1993).
Article CAS Google Scholar - Kopf, M. et al. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature 362, 245–248 (1993).
Article CAS Google Scholar - Muller, U. et al. Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921 (1994).
Article CAS Google Scholar - Kopf, M. et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342 (1994).
Article CAS Google Scholar - Rickert, R.C., Rajewsky, K. & Roes, J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature 376, 352–355 (1995).
Article CAS Google Scholar - Ahearn, J.M. et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity 4, 251–262 (1996).
Article CAS Google Scholar - Lutz, C. et al. IgD can largely substitute for loss of IgM function in B cells. Nature 393, 797–801 (1998).
Article CAS Google Scholar - Wessels, M.R. et al. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA 92, 11490–11494 (1995).
Article CAS Google Scholar - Cascalho, M. et al. A quasi-monoclonal mouse. Science 272, 1649–1652 (1996).
Article CAS Google Scholar - Bachmann, M.F. et al. The role of antibody concentration and avidity in antiviral protection. Science 276, 2024–2027 (1997).
Article CAS Google Scholar - Lieberman, R. et al. Genetics of a new IgVH (T15 idiotype) marker in the mouse regulating natural antibody to phosphorylcholine. J. Exp. Med. 139, 983–1001 (1974).
Article CAS Google Scholar - Oxenius, A., Bachmann, M.F., Zinkernagel, R.M. & Hengartner, H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 28, 390–400 (1998).
Article CAS Google Scholar - Oehen, S.U. et al. Escape of thymocytes and mature T cells from clonal deletion due to limiting tolerogen expression levels. Cell Immunol. 158, 342–352 (1994).
Article CAS Google Scholar - Bachmann, M.F. et al. The influence of antigen organization on B cell responsiveness. Science 262, 1448–1451 (1993).
Article CAS Google Scholar - Banchereau, J. et al. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811 (2000).
Article CAS Google Scholar - Klein, M.A. et al. A crucial role for B cells in neuroinvasive scrapie. Nature 390, 687–690 (1997).
Article CAS Google Scholar - Levy, M., Kleinknecht, C. & Peix, A. Membranous nephropathy and HBs Ag. Lancet 1, 113 (1979).
Article CAS Google Scholar - Casato, M. et al. Cryoglobulinaemia and hepatitis C virus. Lancet 337, 1047–1048 (1991).
Article CAS Google Scholar - Nardi, M., Tomlinson, S., Greco, M.A. & Karpatkin, S. Complement-independent, peroxide-induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell 106, 551–561 (2001).
Article CAS Google Scholar - Choudhury, L.A. et al. Disruption of T cell tolerance to self-immunoglobulin causes polyclonal B cell stimulation followed by inactivation of responding autoreactive T cells. J. Immunol. 164, 1713–1721 (2000).
Article CAS Google Scholar - Wykes, M., Pombo, A., Jenkins, C. & MacPherson, G.G. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 161, 1313–1319 (1998).
CAS PubMed Google Scholar - Sullivan, K.E. Complement deficiency and autoimmunity. Curr. Opin. Pediatr. 10, 600–606 (1998).
Article CAS Google Scholar - Odermatt, B. et al. Virus-triggered acquired immunodeficiency by cytotoxic T-cell-dependent destruction of antigen-presenting cells and lymph follicle structure. Proc. Natl. Acad. Sci. USA 88, 8252–8256 (1991).
Article CAS Google Scholar - Batista, F.D. & Neuberger, M.S. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J. 19, 513–520 (2000).
Article CAS Google Scholar - Plagemann, P.G., Swim, H.E. Propagation of lactic dehydrogenase-elevating virus in cell culture. Proc. Soc. Exp. Biol. Med. 121, 1147–1152 (1966).
Article CAS Google Scholar - Cobbold, S.P. et al. Therapy with monoclonal antibodies by elimination of T cell subsets in vivo. Nature 312, 548–551 (1993).
Article Google Scholar - Ciurea, A. et al. Persistence of lymphocytic choriomeningitis virus at very low levels in immune mice. Proc. Natl. Acad. Sci. USA 96, 11964–11969 (1999).
Article CAS Google Scholar - Ochsenbein, A.F. et al. Protective T cell-independent antiviral antibody responses are dependent on complement. J. Exp. Med. 190, 1165–1174 (1999).
Article CAS Google Scholar - Oxenius, A. et al. Functional in vivo MHC class II loading by endogenously synthesized glycoprotein during viral infection. J. Immunol. 158, 5717–5726 (1997).
CAS PubMed Google Scholar
Acknowledgements
We thank A. Lamarre and P. Klenermann for discussions; E. Niederer, P. Pei and M. Hersberger for help with serum electrophoresis; E. Wacker, A. Probst and A. Zollinger for help with isoelectric focusing; and M. Cascalho for nitrophenyl-specific hybridomas.
Author information
Author notes
- Lukas Hunziker and Mike Recher: These authors contributed equally to this work.
Authors and Affiliations
- Institute for Experimental Immunology, University Hospital, Schmelzbergstrasse 12, Zurich, CH-8091, Switzerland
Lukas Hunziker, Mike Recher, Andrew J. Macpherson, Adrian Ciurea, Stefan Freigang, Hans Hengartner & Rolf M. Zinkernagel - University Hospital Basel, Internal Medicine A, Petersgraben 4, Basel, 4021, Switzerland
Lukas Hunziker
Authors
- Lukas Hunziker
You can also search for this author inPubMed Google Scholar - Mike Recher
You can also search for this author inPubMed Google Scholar - Andrew J. Macpherson
You can also search for this author inPubMed Google Scholar - Adrian Ciurea
You can also search for this author inPubMed Google Scholar - Stefan Freigang
You can also search for this author inPubMed Google Scholar - Hans Hengartner
You can also search for this author inPubMed Google Scholar - Rolf M. Zinkernagel
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toLukas Hunziker or Mike Recher.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Hunziker, L., Recher, M., Macpherson, A. et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections.Nat Immunol 4, 343–349 (2003). https://doi.org/10.1038/ni911
- Received: 03 December 2002
- Accepted: 14 January 2003
- Published: 10 March 2003
- Issue Date: 01 April 2003
- DOI: https://doi.org/10.1038/ni911