Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract (original) (raw)
Main
Respiratory syncytial virus (RSV) is the most common cause of viral lower respiratory tract infection. RSV can cause severe disease in infants1, in older immunodeficient children2 and the elderly3. Reinfection by RSV is a frequent event in infants4,5. Adults also remain susceptible to repeated RSV infection, indicating that protective immunity to reinfection may be incomplete and of short duration6. In murine models, CD8+ T cells have been reported to play a pivotal role in recovery from RSV infection7,8. In BALB/c mice, a single immunodominant, H-2Kd-restricted peptide epitope in the matrix 2 (M2) protein of RSV (M282–90) is the major target of the CD8+ T cells responding to RSV infection; these CD8+ T cells appear to have a prominent role in both recovery from RSV infection and protective immunity9,10. However, the protective immunity conferred by M2-specific CD8+ T cells is short-lived and rapidly wanes11, suggesting that the susceptibility to reinfection may be the result of immune dysregulation mediated by RSV.
To address the issue of possible immunosuppression mediated by RSV, we examined and characterized the dominant M282–90-specific CD8+ T-cell response in the lungs of BALB/c mice during RSV infection using tetramer staining and functional assays. We found that there is a large expansion of activated, M282–90-specific CD8+ T cells in the lungs during the course of primary and challenge RSV infection, indicating that the initial activation and proliferation of M2-specific CD8+ T-cell precursors is normal. However, the massively expanded and activated M282–90-specific CD8+ T-cell population that infiltrated the lungs showed significantly impaired expression of effector activity. Impaired processes included cytokine synthesis and ex vivo cytolytic activity, as well as development of memory/effector cells in the lung. This impaired effector phenotype was due to defective TCR signaling in the effector T cells and required pulmonary RSV infection. These results strongly suggest that RSV infection results in defective TCR-mediated signaling and expression of effector activity via activated lung CD8+ T cells and impaired development of CD8+ T-cell memory.
Lung CD8 + T cells in RSV are deficient in cytokines
We determined the magnitude and the kinetics of the primary CD8+ T-cell response in the lungs of BALB/c mice during experimental intranasal RSV infection. We stained CD8+ T cells infiltrating the lungs with RSV major histocompatibility complex (MHC) class I H-2Kd tetramer complexes loaded with M282–90 (designated as M2Tet). In parallel, we enumerated the frequency of M2-specific effector CD8+ T cells present in the lung by intracellular cytokine staining (ICS). Approximately 41% of the CD8+ T cells infiltrating the lungs of infected animals at the peak of the primary CD8+ response (day 8) stained specifically with the M2Tet (Fig. 1_a_). By contrast, about 20% of the lung CD8+ T cells produced interferon-γ (IFN-γ) in response to the M282–90 peptide. Therefore, only about 50% of the M282–90-specific CD8+ T cells present in the infected lungs (as defined by M2Tet staining) appeared to produce IFN-γ in response to antigen. A similar discrepancy between tetramer staining and cytokine production was also observed for antigen-dependent TNF-α production by lung-infiltrating CD8+ T cells (in this instance, <10% of the expected frequency; data not shown).
Figure 1: Visualization and quantification of M2-specific CD8+ T cells during RSV infection in BALB/c mice.
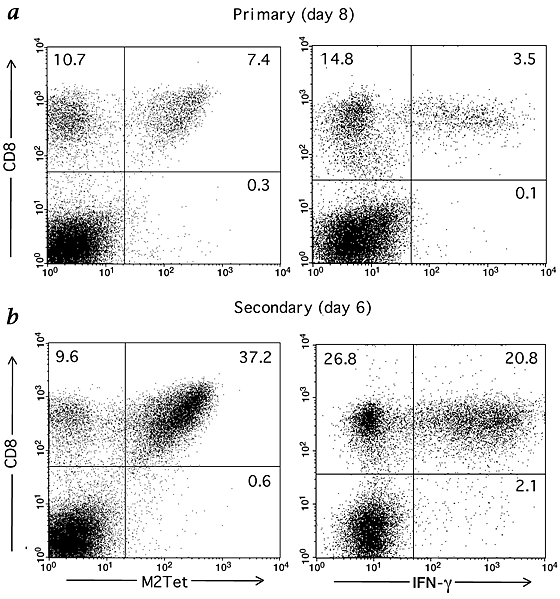
a and b, Lung-derived lymphocytes were prepared from infected animals at day 8 post-infection (a) or from vvM2-primed mice at day 6 post-challenge (b), stained for CD8 and M2Tet tetramer, and analyzed by flow cytometry. The same batches of cells were cultured in the presence of M282–90 peptide, then stained for CD8, followed by staining for intracellular IFN-γ. The numbers given show the percentages of CD8+ T cells that are M2Tet+ or IFN-γ+. Results are representative of several independent experiments in which proportions of M2Tet+CD8+ T cells were consistent.
During the peak of the primary CD8+ T-cell response in the lungs (day 8–12; Table 1), only 40–60% of the expected number of M282–90-specific CD8+ T cells (as defined by M2Tet staining) secreted IFN-γ. This difference in tetramer staining and IFN-γ production was demonstrable as late as 60–90 days after infection when the frequency of M2Tet+CD8+ cells in the lungs had declined to unexpectedly low levels (2–4%). At these later time points, the frequency of residual M2Tet+CD8+ T cells in the lungs that secreted IFN-γ was even lower than the frequency of IFN-γ+CD8+ cells at the peak of the response. Throughout the course of primary RSV infection, more than 90% of the CD8+ T cells infiltrating the lungs expressed the CD11HiCD44HiCD62LLo phenotype, which is characteristic of effector/memory cells throughout the course of primary RSV infection (Table 1). Thus, this deficiency in effector activity was exhibited at least by RSV M2-specific CD8+ T cells localized to this peripheral pulmonary memory/effector T-cell compartment.
Table 1 Kinetics of M282–90-specific CD8+ T-cell response in primary RSV infectiona
We recently identified and characterized a second subdominant MHC class I epitope within the RSV F protein12. F-specific CD8+ T cells responding in the lung to RSV infection also displayed a comparable defect in cytokine synthesis12, suggesting that this deficit in effector activity is not unique to the dominant M2-specific subpopulation of lung CD8+ T cells and is characteristic of the host CD8+ T-cell response to RSV.
This discrepancy between tetramer staining and intracellular IFN-γ production as measures of antigen-specific CD8+ T-cell accumulation in the lung during primary RSV infection was unexpected; in other models of experimental pulmonary virus infection (for example, type A influenza), these two techniques for the quantification of antigen-specific CD8+ cells in the lungs directly correlated13. To ensure that this deficit was not a general property of CD8+ T cells infiltrating the lungs during respiratory virus infection, we examined, in parallel, tetramer staining and intracellular IFN-γ production by CD8+ T cells infiltrating the lungs during primary infection of BALB/c mice with the A/Japan/305/57 influenza virus. At the peak of the primary response to influenza virus in the lungs (day 8), we found that the frequency of tetramer+ and IFN-γ+ lung CD8+ T cells directed to the dominant hemagglutinin (HA)204–212 epitope was comparable (Table 1). The frequency of lung CD8+ T cells directed to the three other major influenza A CD8+ T-cell epitopes14, HA210–219, HA529–537 and nucleoprotein (NP)147–155, at day 8 of infection also showed a close correspondence between tetramer staining and ICS (HA210–219: 4% Tet+, 4% IFN-γ+; HA529–537: 12% Tet+, 13% IFN-γ+; NP147–155: 17% Tet+, 22% IFN-γ+). This was maintained throughout pulmonary influenza virus infection (data not shown).
RSV-specific CD8 + T cells show impaired effector activity
We next asked whether a similar defect in effector activity is detectable in the response of memory CD8+ T cells accumulating in the lungs after a challenge RSV infection. To enrich for responding M282–90-specific T cells, we primed mice with a recombinant vaccinia virus expressing the RSV M2, followed by a challenge intranasal infection with RSV three to four weeks after priming. We15 and others16 have previously reported that this priming strategy leads to a robust in vitro secondary CTL response from immune splenocytes to infectious RSV. At day 6 after challenge infection, approximately 80% of the CD8+ T cells stained with the M2Tet whereas only about 43% of the CD8+ cells produced IFN-γ in response to antigen (that is, ∼53% of the expected percentage of M2-specific cells based on tetramer staining; Fig. 1_b_). Kinetic analysis revealed the expected, accelerated memory response to M2 as demonstrated by the rapid accumulation of the M2Tet+CD8+ T cells as early as day 4 after challenge (Table 2). The percentage of M2Tet+CD8+ cells in the lungs peaked at day 6 (∼79%) and was maintained at a high level through post-challenge day 12. However, as in primary RSV infection, the frequency of cells producing IFN-γ in response to the M282–90 peptide during the peak of the memory response ranged from 52 to 66% of the expected percentage based on tetramer staining. Although the frequency of M2Tet+CD8+ T cells remained relatively high long after challenge RSV infection (day 45), the percentage of the M2-specific CD8+ T cells in the lungs producing IFN-γ in response to antigen dropped disproportionately. This again suggests that, as in the primary response, RSV infection alters the establishment and maintenance of peripheral CD8+ T-cell memory in the lung compartment.
Table 2 Kinetics of M282–90-specific CD8+ T-cell response in secondary RSV infection
We considered the possibility that these activated IFN-γ−CD8+ T cells infiltrating the lungs during primary and challenge infection belonged to the Type 2 cytokine subset17. The Type 2 cytokines interleukin-4 (IL-4), IL-5 and IL-13 were not detected by ELISPOT or ELISA in response to specific peptide stimulation (data not shown).
RSV selectively impairs CD8 + T-cell effector activity in lungs
The recruitment of many RSV M2-specific CD8+ T cells to the lung during both primary and challenge RSV infection suggested that the activation and proliferation of virus-specific CD8+ T cells in secondary lymphoid tissues were unaffected by RSV. To substantiate this point, we first compared the frequency of resident RSV-specific M2Tet+ and IFN-γ+CD8+ T cells found in the spleens of RSV-infected animals during primary infection. Although the frequencies of M2-specific CD8+ T cells present in the spleen defined by either tetramer staining or antigen-dependent cytokine production were low (typically <6% of total splenic CD8+ T-cell numbers), over the course of primary infection, the estimated frequency of M2-specific CD8+ T cells determined by either method was comparable; IFN-γ staining reproducibly yielded slightly higher frequency estimates (Fig. 2_a_). This result suggested that, in contrast to the lungs, there was no impairment in the response of CD8+ T effector cells or memory cells to RSV in this central lymphoid compartment.
Figure 2: Effector activity, antigen-dependant TCR downregulation and IL-2 rescue of M2-specific lung and spleen CD8+ T cells.
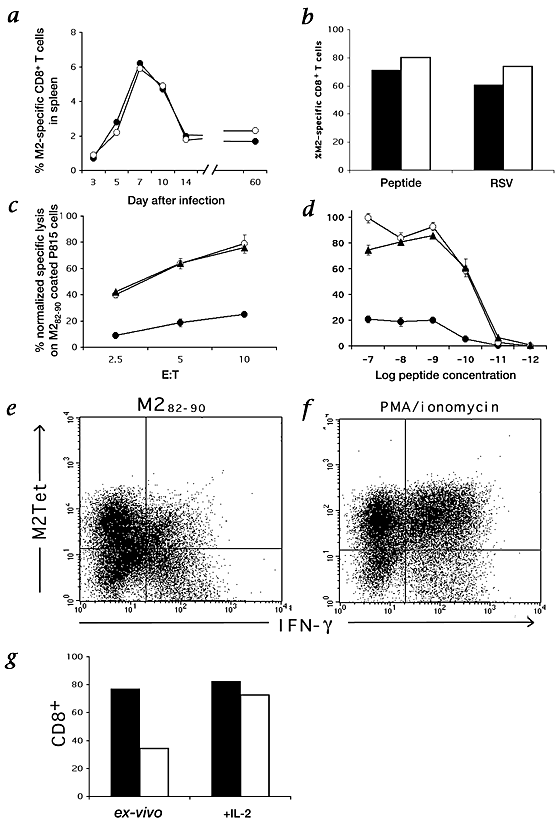
a, Spleen cells were isolated at the indicated times after primary intranasal RSV infection and where CD8+ cells stained for M2Tet (●) and independently for intracellular IFN-γ synthesis (○) by flow cytometry. b, _In vitro_-stimulated splenocytes were derived from pooled spleens of vvM2-immunized mice, stimulated with irradiated naive splenocytes pulsed with 5 × 10−8M M282–90 peptide or infected with RSV at MOI of 1. Cells were cultured for 6 d in the presence of IL-2 (10 U/ml), and analyzed for M2Tet staining (▪) and intracellular IFN-γ synthesis (□). c and d, Lung cells derived from vvM2-primed and RSV-challenged mice (●) and _in vitro_-cultured splenocytes (▴, peptide-stimulated; ○ RSV-stimulated) at the peak of antigen-specific response (day 6 post-challenge and post-stimulation, respectively) were assayed at the indicated E:T (c) ratio and peptide concentration (d) in a standard 51Cr release assay. Specific lysis values were normalized to the actual number of M2Tet+ cells in each population. Results are representative of 4 independent experiments. e and f, Down-modulation of TCR after cognate peptide stimulation. Lung lymphocytes derived from vaccinia M2-primed and RSV-challenged mice at day 6 post-infection were incubated in the presence of M282–90 peptide (e; 1 × 10−6M) or PMA/ionomycin (f). Cells were stained for CD8 and M2Tet, permeabilized, and then stained for intracellular IFN-γ. Plots are gated on CD8+ cells, and numbers represent the percentage of gated cells in each quadrant. g, Day 6 lung CD8+ T cells from RSV-challenged mice were purified using magnetic beads and cultured with IL-2 for 12–18 h. Cells were then analyzed for M2Tet staining (▪) and IFN-γ production (□).
To further explore this issue, we examined the IFN-γ response of immune M2-specific CD8+ T-cell effectors at day 6 after in vitro stimulation of splenocytes from M2-immune donors with RSV-infected or M282–90-pulsed splenic antigen-presenting cells (APCs). We compared these responses with that of lung CD8+ T cells isolated 6 days after challenge RSV infection of M2-primed donors. Cultures of M2-immune splenocytes stimulated with either RSV-infected cells or peptide were enriched for M2Tet+CD8+ T cells, which were present at frequencies comparable to those detected in the RSV-infected lungs (Fig. 2_b_). However, unlike lung-derived CD8+ T cells, activated, M2-specific, spleen-derived effector CD8+ T cells showed no deficit in IFN-γ synthesis. The cytokine response and cytolytic activity (see below) of effector CD8+ T cells generated from immune spleens in response to RSV-infected APC were identical with or without IL-2 supplementation to the culture medium during effector T-cell generation in vitro (data not shown).
We next investigated whether the activated lung CD8+ T cells also impaired ex vivo cytolytic activity. CD8+ T cells were isolated from the lungs of M2-primed mice 6 days after challenge RSV infection and examined directly for ex vivo cytolytic activity on M282–90-pulsed targets cells. In parallel, we analyzed M2-specific cytolytic activity of activated CD8+ T cells generated in vitro from the spleen of M2-immune donors 6 days after stimulation with RSV-infected or M282–90-pulsed APCs. Effector:target (E:T) ratios were normalized based on the frequency of M2Tet+CD8+ T cells in each effector population. As reported10,15, effectors cells generated in vitro in response to infectious RSV or M2 peptide displayed potent cytolytic activity over a range of E:T ratios (Fig. 2_c_). By contrast, M2-specific CD8+ T cells from the infected lungs routinely displayed less than 25% of the activity of spleen-derived effectors over the same E:T ratios (Fig. 2_c_). The peptide concentration for optimal lysis (1 × 10−7 to 1 × 10−9 M) was identical for lung- and spleen-derived effector cells (Fig. 2_d_), suggesting that there was not selective recruitment of low-avidity effector cells to the lungs during RSV infection. The peptide dose-dependence of IFN-γ synthesis by ICS for lung and spleen CD8+ T-cell effectors directly paralleled the peptide dose dependence of in vitro cytolytic activity (data not shown).
Defective lung CD8 + T cells are impaired in TCR signaling
To further explore the basis for the weak cytolytic activity of lung-derived M2-specific CD8+ T cells, we analyzed the intracellular perforin content of lung- and spleen-derived effector T cells from M2-immune donors. Using flow cytometry with anti-perforin antibody, we found, in contrast to published observations18, that the intracellular perforin content of both the ex vivo lung-derived (Fig. 3_a_) and in vitro spleen-derived (Fig. 3_b_) CD8+ T-cell effectors was low. However, in companion studies, we observed that the short-term cocultivation of activated effector CD8+ T cells with specific peptide under conditions resulting in IFN-γ synthesis, simultaneously resulted in the upregulation of perforin expression by the T cells (our unpublished observations).
Figure 3: Perforin expression of M2-specific cells derived from the lung and in vitro stimulated splenocytes culture.
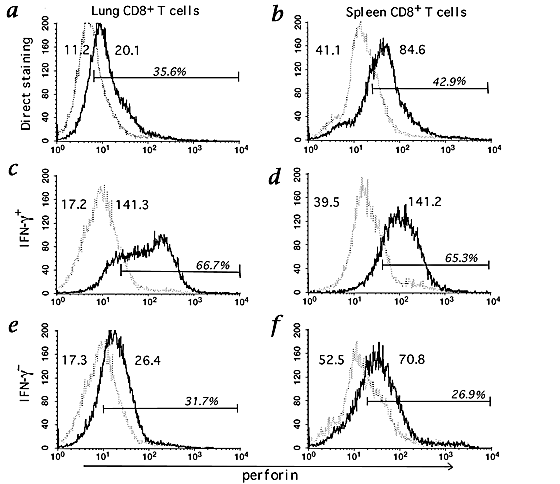
a and b, Lung CD8+ T cells at day 6 post-challenge with RSV and splenic CD8+ T cells at day 6 after M282–90 peptide stimulation were directly stained with anti-perforin (solid line) or isotype control antibody (dotted line). c–f, The same batches were treated as in intracellular IFN-γ assay, permeabilized, and then stained for CD8, intracellular perforin, and IFN-γ. Cells were gated on IFN-γ+CD8+ (c and d) or IFN-γ−CD8+ cells (e and f). The numbers given are mean fluorescence intensity for each staining and bar shows the percentage of stained cells above isotype control staining.
We next determined if the weak cytolytic activity of lung-derived CD8+ T-cell effectors might be linked to the inability of the IFN-γ− fraction of M2-specific T cells to upregulate perforin expression in response to the antigen. We purified CD8+ cells from the lungs of M2-primed mice (day 6 post-challenge RSV infection) and from cultures of M2-immune splenocytes 6 days after in vitro stimulation with RSV or M282–90 peptide. These CD8+ T-cell populations were exposed to specific peptide in short-term culture (5 h at 37 °C) and then analyzed for IFN-γ production and perforin expression. The IFN-γ+ and IFN-γ−CD8+ T cells were separately gated, and the intracellular perforin content of the gated cell populations was determined. Splenic, M2-specific, IFN-γ+ T cells displayed uniform upregulation of perforin expression in response to peptide (Fig. 3_d_). The small fraction (<15%) of splenic CD8+ T cells that were IFN-γ− did not upregulate perforin (Fig. 3_f_) and likely represent residual irrelevant viable CD8+ T cells. The IFN-γ+CD8+ T cells isolated from RSV-infected lungs also upregulated perforin expression (Fig. 3_c_). By contrast, lung IFN-γ−CD8+ T cells which consisted primarily of the M2Tet+ cells (approximately 70% of the total IFN-γ−CD8+ T-cell population in the infected lungs) failed to upregulate perforin expression (Fig. 3_e_).
Activated CD8+ T cells typically down-modulate cell-surface T-cell receptor (TCR) expression in response to antigen19, resulting in a concomitant loss of specific tetramer staining. To directly assess if CD8+ T cells responding to RSV infection in the lung were defective in antigen-dependent TCR down-modulation, we analyzed M2Tet staining and IFN-γ production by activated lung CD8+ T cells from M2-primed mice undergoing challenge RSV infection after short-term in vitro exposure to the M2 peptide. As Fig. 2_e_ demonstrates, the majority (>60%) of the total lung M2-specific CD8+ T cells (defined by either M2Tet staining or IFN-γ production) did not secrete IFN-γ in response to peptide and likewise showed no decrease in M2Tet staining. This impairment in expression of effector activity by lung CD8+ T cells was not overcome even following in vitro exposure to phorbol 12-myristate 13-acetate (PMA)/ionomycin; that is, less than 60% of lung M2Tet+ T cells secrete IFN-γ in response to this non-specific mitogenic stimulus (Fig. 2_f_).
The unresponsiveness of RSV-specific lung CD8+ T cells to antigenic stimulation is reminiscent of the refractory state observed in anergic T cells20. Because T-cell anergy can be reversed by exposure of the cells to IL-2 (ref. 21), we questioned whether the defect in TCR signaling exhibited by RSV-specific lung CD8+ T cells might likewise be reversed by IL-2 treatment. To test this possibility, CD8+ T cells were purified from the lungs of M2-immune mice six days after challenge RSV infection. Approximately 80% of the purified T cells were M2-specific as judged by direct ex vivo tetramer staining and 40–50% of the lung CD8+ T cells secreted IFN-γ in response to peptide stimulation. Purified lung CD8+ T cells were cultured overnight (12–18 h at 37 °C) in medium with or without IL-2 supplementation (10 U/ml), and then tested for IFN-γ production by ICS in response to M2 peptide stimulation. Lung CD8+ T cells incubated overnight in medium without IL-2 showed a 50% drop in overall cell viability and only 25–30% of the remaining viable T cells produced IFN-γ in response to the peptide (Fig. 2_g_). By contrast, lung CD8+ T cells exposed to IL-2 showed no loss in cell viability and no increase in total cell counts (cell yields were ∼100–105% of starting cell numbers). When IL-2-treated CD8+ T cells were evaluated for IFN-γ production in response to M2 peptide, more than 80% of the predicted number of M2-specific CD8+ T cells (based on tetramer staining) were IFN-γ+ (Fig. 2_g_). These data suggest that the block in antigen-dependent TCR-mediated signaling had been reversed.
Defective CD8 + T-cell response requires pulmonary infection
Our results show expansion and accumulation of M2-specific CD8+ T cells in the lungs during primary and challenge RSV infection, as well as normal effector activity displayed by resident spleen CD8+ T cells during RSV infection. These data suggest that the response to RSV infection was unimpaired in central lymphoid tissue and that pulmonary RSV infection might be critical for the development of impaired effector activity displayed by lung-infiltrating CD8+ T cells. If so, then M2-specific CD8+ T cells recruited to the lungs in response to M2 antigen without concomitant RSV infection might display a normal effector T-cell phenotype. To test this, we made use of the recent finding that intranasal introduction of plasmid DNA encoding specific antigens under control of a strong promoter stimulates the development of an antigen-specific cellular immune response in the lungs22,23. We therefore introduced plasmid DNA encoding the full-length RSV M2 protein intranasally into M2-primed mice; we did this in conjunction with CpG oligodeoxynucleotides (ODNs), which stimulate local inflammation and enhance CD8+ T-cell recruitment to the lungs24. At day 6 after DNA immunization, the frequency of M2Tet+CD8+ T cells infiltrating the lungs of plasmid recipients (∼13%) was lower than that of mice undergoing challenge RSV infection. However, about 96% of the expected number of M2-specific CD8+ T cells in the lungs of DNA challenged mice scored positive for IFN-γ production by ICS (Table 3).
Table 3 T-cell cytokine response to RSV and DNA challenge
Discussion
Here we analyzed the CD8+ T-cell responses in the lungs to the immunodominant M282–90 epitope during primary and challenge RSV infection. Pulmonary RSV infection induces a massive expansion and accumulation of M282–90-specific CD8+ T cells in the lungs. However, although these M2Tet+ T cells displayed the phenotype of activated effectors and peripheral memory/effector cells, they were deficient in both pro-inflammatory cytokine secretion and ex vivo cytolytic activity. Furthermore, the frequency of RSV M2-specific memory CD8+ T cells in the lung compartment rapidly declined after recovery from primary RSV infection, and the cytokine response of the RSV-specific memory CD8+ T cells was significantly reduced late after challenge infection.
This selective impairment in lung CD8+ T-cell responsiveness induced by RSV likely reflects a defect in antigen-dependent TCR-mediated signaling. Along with the deficiency in cytokine synthesis and ex vivo cytolytic activity, M2Tet+IFN-γ− lung CD8+ T-cell effectors also failed to upregulate perforin and down-modulate cell-surface TCRs in response to antigen. Reversal of the block in antigen-dependent cytokine synthesis by IL-2 is noteworthy and directly parallels the restoration of antigen responsiveness observed in anergic T cells after IL-2 treatment21,25; in those cells, alterations or deficiencies in intracellular signaling intermediates involved in TCR-dependent signal transduction have been implicated in the defective antigen responsiveness of anergic T cells21,26. The refractory RSV-specific lung CD8+ T-cell effectors may likewise have an alteration in one or more intracellular signaling intermediates necessary for TCR-dependant triggering of effector activity which can be overcome by IL-2 receptor engagement21.
Although pulmonary RSV infection does alter peripheral (lung) CD8+ effector activity and memory T cells, it does not affect the initial activation and expansion of primary or memory CD8+ T-cell precursors or the recruitment of activated CD8+ T cells from central lymphoid tissues to the lungs. Nor does RSV infection affect the effector activity, frequency or function of central (splenic) effector/memory CD8+ T cells. Given that lung M2-specific CD8+ T cells responding to intranasal plasmid DNA immunization likewise show no deficit in effector activity, RSV replication in the respiratory tract may be required for the development of the impaired CD8+ T-cell effectors and memory response. Respiratory epithelial cells are the primary targets of productive RSV replication27, whereas macrophages and dendritic cells are abortively infected28. Productive RSV infection in the respiratory tract could lead to the selective inhibition of lung CD8+ T-cell function, either directly through the accumulation of one or more RSV gene products capable of suppressing host immune/inflammatory responses (for example, RSV G and/or SH)29 or indirectly through the releases of inhibitory factors from RSV-infected respiratory epithelial cells30,31,32. In either case, RSV-induced inhibition of CD8+ T-cell effector activity and memory development and maintenance would be restricted to a respiratory tract where RSV gene products and productively infected epithelial cells are abundant.
An intriguing feature of RSV infection is the susceptibility of previously infected individuals to reinfection with antigenically closely related viruses or the identical virus strain. In infants, reinfection with RSV has been reported to occur within weeks of recovery from primary infection33. In adults with high levels of circulatory neutralizing antibodies, upwards of 25% immune individuals could be re-infected with RSV of the same serotype within two months of natural infection6. These and related observations suggest that the duration of protective immunity to challenge RSV infection is short-lived. The results reported here suggest one possible mechanism for the absence of durable long-lived immunity to RSV infection: Perhaps through its effect on TCR-dependent signaling, RSV suppresses the development of peripheral CD8+ T-cell memory in the lungs. If RSV also inhibits the function of virus-specific CD4+ T-cell effectors in the lungs, then RSV infection could impact on the strength and durability of both cellular and humoral immunologic memory in this compartment. Finally, our results suggest that a localized respiratory infection with an immunosuppressive virus like RSV can inhibit the function of effector T cells and local immunologic memory development at that peripheral site without impacting on virus-specific T-cell activation or memory development in central lymphoid tissues.
Methods
Mice.
6–8-wk-old female BALB/c mice (H-2d) were purchased from Taconic Farms Inc. (Germantown, New York). Mice were housed in a pathogen-free environment in an AAALAC-approved vivarium at the University of Virginia.
Viruses and infection of mice.
The A2 strain of RSV was a gift from P.L. Collins. RSV stock was grown on HEp-2 cells (ATCC, Manassas, Virginia) and titered for infectivity. vvM2 was a gift from J.L. Beeler. Mice were infected with 5 × 106 plaque-forming units (p.f.u.) of vvM2 by scarification at the base of tail. 3–4 wk after priming, mice were lightly anesthetized with 2:1 mixture of ether and chloroform, and intranasally inoculated with 1 × 106 p.f.u. of RSV in 50 μl. At various times after infection, infected mice were killed by cervical dislocation.
Preparation and culture of lymphocytes.
Lungs were perfused with 5 ml of PBS containing 10 U/ml heparin (Sigma) through the right ventricle using a syringe fitted with 25-gauge needle. The lungs and spleens were removed and placed into Iscove's modified Dulbecco's medium (IMDM) supplemented with glutamine, gentamicin, penicillin G and 10% FBS. The tissue was processed through a steel screen to obtain single-cell suspension and particulate matter was removed by quick centrifugation at 1000_g_. Cells were counted and resuspended at the given cell concentrations for the appropriate in vitro assay. Pooled spleen cells from vvM2-immunized mice were resuspended in complete IMDM supplemented with recombinant human IL-2 (20 U/ml), and 2.5 × 107 cells were added to each well of a 6-well plate. Syngeneic splenocyte stimulators were prepared from naive mice by irradiation (2000 rad) and pulsing for 0.5 h at room temperature with the peptide or infecting with RSV for 1 h at 37 °C at multiplicity of infection (MOI) of 1. Thereafter, cells were extensively washed to remove free unbound peptide or virus, and 5 × 106 cells were added to the each well of responder cells. In companion experiments, immune splenocytes were cultured with irradiated, RSV-infected splenocytes in medium without supplemental IL-2. The effector T cells that were generated exhibited the same cytokine response and cytolytic activity as did effector CD8+ T cells that were generated in the presence of IL-2 in culture medium. To purify CD8+ cells, cell suspensions were positively selected with mouse CD8 microbeads and MACS separation column (Miltenyi Biotec, Auburn, California) according to manufacturer's instructions. The typical purification results in 80–90% CD8+ cell purity.
Flow cytometry and tetramer staining.
MHC class I–peptide tetramers were produced as described34. Freshly explanted lung lymphocytes or in vitro cultured splenocytes (1 × 106) were purified by density-gradient centrifugation and stained in PBS:3% (wt:vol), FBS:0.09% (wt:vol) NaN3 using fluorochrome-conjugated antibodies and MHC class I tetramers. Antibodies used were anti-CD8 (clone 53-6.7), anti-CD11a (clone 2D7), anti-CD44 (clone IM7) and anti-CD62L (clone MEL-14). All antibodies were purchased from PharMingen (San Diego, California). After staining, cells were fixed in PBS:2% (wt:vol) paraformaldehyde, and events acquired using a FACSCalibur flow cytometer (Becton Dickinson, San Diego, California). Dead cells were excluded on the basis of forward and side light scatter. Data were analyzed using CELLQuest (Becton Dickinson).
Intracellular staining.
To enumerate the number of cytokine-producing cells, intracellular cytokine staining was performed as previously35. In brief, 1 × 106 freshly explanted lung lymphocytes were cultured in culture tube. Cells were left untreated, stimulated with M282–90 peptide, or treated with PMA (50 ng/ml) and ionomycin (500 ng/ml). In certain experiments using MACS-purified lung CD8+ cells or in vitro cultured splenocytes, 5 × 105 P815 cells were added as APCs. In all cases cells were incubated for 5 h at 37 °C in 7% CO2. Brefeldin A (10 μg/ml; Sigma) was added for the duration of the culture period to facilitate intracellular cytokine accumulation. The antibodies used were anti-IFN-γ (clone XMG1.2) or its control isotype antibody (rat IgG1). Intracellular perforin staining was performed as described18.
CTL assay.
A standard chromium release assay was performed as described15. Effectors from lung-derived lymphocytes or _in vitro_-cultured splenocytes were prepared by density gradient centrifugation. For peptide titration, assays were performed at a constant E:T ratio of approximately 2.5:1. Percent specific lysis was calculated as follows: [(51Cr release with effector cells – spontaneous 51Cr release) ÷ (total 51Cr release with 1% Triton X-100 – spontaneous 51Cr release)] × 100. The percent specific-lysis values represent the mean values of quadruplicate wells.
Intranasal plasmid DNA injection.
The plasmid, pCI-neo-M2, containing M2 gene of RSV under the control of cytomegalovirus immediate early promoter was constructed by inserting M2 gene fragment into _Xho_I and _Not_I sites of pCI-neo (Promega, Madison, Wisconsin). LPS-free DNA was prepared using EndoFree Plasmid Mega kit (Qiagen, Chatsworth, California) according to the manufacturer's instruction, and 100 μg of plasmid with 10 μg CpG ODN was injected intranasally to the left nostril of mice in a volume of 100 μl. pCI-neo parental vector plasmid was used as a control.
References
- Glezen, P. & Denny, F.W. Epidemiology of acute lower respiratory disease in children. N. Engl. J. Med. 288, 498–505 (1973).
Article CAS PubMed Google Scholar - Chanock, R.M., Parrott, R.H., Connors, M., Collins, P.L. & Murphy, B.R. Serious respiratory tract disease caused by respiratory syncytial virus: prospects for improved therapy and effective immunization. Pediatrics 90, 137–143 (1992).
CAS PubMed Google Scholar - Falsey, A.R. & Walsh, E.E. Relationship of serum antibody to risk of respiratory syncytial virus infection in elderly adults. J. Infect. Dis. 177, 463–466 (1998).
Article CAS PubMed Google Scholar - Glezen, W.P., Taber, L.H., Frank, A.L. & Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child 140, 543–546 (1986).
CAS PubMed Google Scholar - Beem, M. Repeated infections with respiratory syncytial virus. J. Immunol. 98, 1115–1122 (1967).
CAS PubMed Google Scholar - Hall, C.B., Walsh, E.E., Long, C.E. & Schnabel, K.C. Immunity to and frequency of reinfection with respiratory syncytial virus. J. Infect. Dis. 163, 693–698 (1991).
Article CAS PubMed Google Scholar - Alwan, W.H., Record, F.M. & Openshaw, P.J. CD4+ T-cells clear virus but augment disease in mice infected with respiratory syncytial virus. Comparison with the effects of CD8+ T-cells. Clin. Exp. Immunol. 88, 527–536 (1992).
Article CAS PubMed PubMed Central Google Scholar - Cannon, M.J., Openshaw, P.J. & Askonas, B.A. Cytotoxic T-cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 168, 1163–1168 (1988).
Article CAS PubMed Google Scholar - Kulkarni, A.B. et al. Cytotoxic T-cells specific for a single peptide on the M2 protein of respiratory syncytial virus are the sole mediators of resistance induced by immunization with M2 encoded by a recombinant vaccinia virus. J. Virol. 69, 1261–1264 (1995).
CAS PubMed PubMed Central Google Scholar - Openshaw, P.J., Anderson, K., Wertz, G.W. & Askonas, B.A. The 22,000-kilodalton protein of respiratory syncytial virus is a major target for Kd-restricted cytotoxic T lymphocytes from mice primed by infection. J. Virol. 64, 1683–1689 (1990).
CAS PubMed PubMed Central Google Scholar - Kulkarni, A.B., Connors, M., Firestone, C.Y., Morse, H.C. & Murphy, B.R. The cytolytic activity of pulmonary CD8+ lymphocytes, induced by infection with a vaccinia virus recombinant expressing the M2 protein of respiratory syncytial virus (RSV), correlates with resistance to RSV infection in mice. J. Virol. 67, 1044–1049 (1993).
CAS PubMed PubMed Central Google Scholar - Chang, J., Srikiatkhachorn, A. & Braciale, T.J. Visualization and characterization of respiratory syncytial virus F-specific CD8(+) T-cells during experimental virus infection. J. Immunol. 167, 4254–4260. (2001).
Article CAS PubMed Google Scholar - Flynn, K.J. et al. Virus-specific CD8+ T-cells in primary and secondary influenza pneumonia. Immunity 8, 683–691 (1998).
Article CAS PubMed Google Scholar - Sweetser, M.T., Braciale, V.L. & Braciale, T.J. Class I major histocompatibility complex-restricted T lymphocyte recognition of the influenza hemagglutinin. Overlap between class I cytotoxic T lymphocytes and antibody sites. J. Exp. Med. 170, 1357–1368 (1989).
Article CAS PubMed Google Scholar - Srikiatkhachorn, A. & Braciale, T.J. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J. Exp. Med. 186, 421–432 (1997).
Article CAS PubMed PubMed Central Google Scholar - Alwan, W.H., Kozlowska, W.J. & Openshaw, P.J. Distinct types of lung disease caused by functional subsets of antiviral T-cells. J. Exp. Med. 179, 81–89 (1994).
Article CAS PubMed Google Scholar - Sad, S., Marcotte, R. & Mosmann, T.R. Cytokine-induced differentiation of precursor mouse CD8+ T-cells into cytotoxic CD8+ T-cells secreting Th1 or Th2 cytokines. Immunity 2, 271–279 (1995).
Article CAS PubMed Google Scholar - Slifka, M.K., Rodriguez, F. & Whitton, J.L. Rapid on/off cycling of cytokine production by virus-specific CD8+ T-cells. Nature 401, 76–79 (1999).
Article CAS PubMed Google Scholar - Valitutti, S., Muller, S., Cella, M., Padovan, E. & Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 375, 148–1451 (1995).
Article CAS PubMed Google Scholar - Zajac, A.J. et al. Viral immune evasion due to persistence of activated T-cells without effector function. J. Exp. Med. 188, 2205–2213 (1998).
Article CAS PubMed PubMed Central Google Scholar - Madrenas, J., Schwartz, R.H. & Germain, R.N. Interleukin 2 production, not the pattern of early T-cell antigen receptor-dependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc. Natl. Acad. Sci. USA 93, 9736–9741 (1996).
Article CAS PubMed PubMed Central Google Scholar - McCluskie, M.J. et al. Direct gene transfer to the respiratory tract of mice with pure plasmid and lipid-formulated DNA. Antisense Nucleic Acid Drug Dev. 8, 401–414 (1998).
Article CAS PubMed Google Scholar - McCluskie, M.J. et al. Route and method of delivery of DNA vaccine influence immune responses in mice and non-human primates. Mol. Med. 5, 287–300 (1999).
Article CAS PubMed PubMed Central Google Scholar - Schwartz, D.A. et al. CpG motifs in bacterial DNA cause inflammation in the lower respiratory tract. J. Clin. Invest. 100, 68–73 (1997).
Article CAS PubMed PubMed Central Google Scholar - Boussiotis, V.A. et al. Prevention of T-cell anergy by signaling through the γc chain of the IL-2 receptor. Science 266, 1039–1042 (1994).
Article CAS PubMed Google Scholar - Sloan-Lancaster, J., Shaw, A.S., Rothbard, J.B. & Allen, P.M. Partial T-cell signaling: Altered phospho-ζ and lack of zap70 recruitment in APL-induced T-cell anergy. Cell 79, 913–922 (1994).
Article CAS PubMed Google Scholar - Hall, C.B., Douglas, R.G., Jr., Schnabel, K.C. & Geiman, J.M. Infectivity of respiratory syncytial virus by various routes of inoculation. Infect. Immun. 33, 779–783 (1981).
CAS PubMed PubMed Central Google Scholar - Becker, S., Quay, J. & Soukup, J. Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J. Immunol. 147, 4307–4312 (1991).
CAS PubMed Google Scholar - Tripp, R.A. et al. Respiratory syncytial virus G and/or SH protein alters Th1 cytokines, natural killer cells, and neutrophils responding to pulmonary infection in BALB/c mice. J. Virol. 73, 7099–7107 (1999).
CAS PubMed PubMed Central Google Scholar - Thomas, L.H., Wickremasinghe, M.I., Sharland, M. & Friedland, J.S. Synergistic upregulation of interleukin-8 secretion from pulmonary epithelial cells by direct and monocyte-dependent effects of respiratory syncytial virus infection. J. Virol. 74, 8425–8433 (2000).
Article CAS PubMed PubMed Central Google Scholar - Preston, F.M., Beier, P.L. & Pope, J.H. Identification of the respiratory syncytial virus-induced immunosuppressive factor produced by human peripheral blood mononuclear cells in vitro as interferon-α. J. Infect. Dis. 172, 919–926 (1995).
Article CAS PubMed Google Scholar - Olszewska-Pazdrak, B. et al. Cell-specific expression of RANTES, MCP-1, and MIP-1α by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 72, 4756–4764 (1998).
CAS PubMed PubMed Central Google Scholar - Henderson, F.W., Collier, A.M., Clyde, W.A. & Denny, F.W. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N. Engl. J. Med. 300, 530–534 (1979).
Article CAS PubMed Google Scholar - Altman, J.D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–6 (1996).
Article CAS PubMed Google Scholar - Murali-Krishna, K. et al. Counting antigen-specific CD8 T-cells: A reevaluation of bystander activation during viral infection. Immunity 8, 177–187 (1998).
Article CAS PubMed Google Scholar
Acknowledgements
We wish to acknowledge the expert and dedicated support of S. Gill in the completion of this work. The work was supported by USPHS grants to T.J.B.
Author information
Authors and Affiliations
- Beirne B. Carter Center for Immunology Research, University of Virginia Health Sciences Center, Charlottesville, Virginia, USA
Jun Chang & Thomas J. Braciale - Department of Pathology and Microbiology, University of Virginia Health Sciences Center, Charlottesville, Virginia, USA
Thomas J. Braciale
Authors
- Jun Chang
You can also search for this author inPubMed Google Scholar - Thomas J. Braciale
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toThomas J. Braciale.
Rights and permissions
About this article
Cite this article
Chang, J., Braciale, T. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract.Nat Med 8, 54–60 (2002). https://doi.org/10.1038/nm0102-54
- Received: 04 September 2001
- Accepted: 03 December 2001
- Issue Date: 01 January 2002
- DOI: https://doi.org/10.1038/nm0102-54