Antiamnesic and Neuroprotective Effects of the Aminotetrahydrofuran Derivative ANAVEX1-41 Against Amyloid β25–35-Induced Toxicity in Mice (original) (raw)
INTRODUCTION
Alzheimer disease (AD) is an irreversible, progressive and degenerative disorder damaging the higher structures of the brain (Selkoe, 1989, 2004). It is actually incurable, as the available treatments, acetylcholinesterase inhibitors or a N_-methyl-D-aspartate receptor antagonist with neuroprotective potential, memantine, are mainly symptomatic. The pathological cleavage of amyloid precursor protein (APP) is responsible for the accumulation of amyloid-β (A_β) proteins, aggregating into fibrillar oligomers and generating amyloid deposits that, in turn, form the senile plaques (Selkoe, 1989, 2004). Oligomers of A_β_ peptides are considered as the main factor mediating the devastating neurotoxicity observed in AD. A_β_ peptides vary in length from 40 to 43 amino acids, A_β_1–42 occurring more frequently and forms fibrillar aggregates far more readily than A_β_1–40 or A_β_1–43 (Selkoe, 1989). Minor fragments were also identified including the highly toxic A_β_25–35 peptide (Kubo et al, 2002; Gruden et al, 2007). The A_β_-mediated toxicity follows a very complex pattern. A_β_ oligomers form Ca2+ permeable pores on plasma membranes and interact with intracellular organelles regulating calcium homeostasis, the endoplasmic reticulum (ER) and mitochondria (Abramov et al, 2004), provoking a massive oxidative stress and induction of neuronal apoptotic death. A_β_ proteins are also responsible for a generalized inflammatory response in brain structures associated with production of cytokines by activated astroglia and microglia (Frederickson, 1992) and exacerbated excitotoxic processes (Mattson et al, 1992).
Moreover, toxicity of A_β_ has recently been shown to be highly dependent on the aggregation species (Chafekar et al, 2008). A_β_ can exist in different assembly states and apart from the monomeric and mature fibrillar stages, different intermediate species have been identified, such as low molecular weight oligomers, larger globular oligomers, and protofibrils. This is true for A_β_1–40 or A_β_1–42/3 but also for A_β_25–35 peptide. These different species greatly differ in their neurotoxic potential and molecular mechanism mediating the toxicity. For instance, impairment of long-term potentiation (Walsh et al, 2002) and ER stress (Chafekar et al, 2007) may be mediated by small oligomers, whereas the neuroinflammatory response may rather involve fibrillar A_β_ (Eikelenboom et al, 2002). Preliminary observations of the laboratory showed that after in vitro aggregation, A_β_25–35 peptide exist in these different species including small oligomers, amorphous oligomers, and fibrillar forms (S Marchal, L Givalois, T Maurice, unpublished work).
We described the nontransgenic model of AD induced in rodents by injection into the lateral ventricle of aggregated A_β_25–35 peptide (Maurice et al, 1996; Delobette et al, 1997). The morphological and biochemical characterization of amyloid toxicity induced by A_β_25–35 has been subsequently analyzed in details. A_β_25–35 induces brain inflammation, oxidative stress, activation of proapoptotic caspases, impairment of long-term potentiation, cell loss in the hippocampus, and memory impairments (Stepanichev et al, 2004, 2006; Meunier et al, 2006). Recently, it was also observed that A_β_25–35 injection activates the glycogen synthase kinase-3_β_, involved in cell survival regulation, T-phosphorylation and APP processing, suggesting that acute A_β_25–35 injection results in production and seeding of endogenous A_β_1–40/42 and T-phosphorylation (Klementiev et al, 2007). The model therefore appears as highly suitable to analyze the putative antiamnesic and neuroprotective activity of drugs with potential interest in AD, as recently used by several authors (Fang and Liu, 2006; Kuboyama et al, 2006; Meunier et al, 2006; Um et al, 2006; Alkam et al, 2007).
The _σ_1 protein has only recently been identified as a chaperone protein located on membranes forming focal contacts between the ER and mitochondria (Hayashi and Su, 2007). In basal conditions, the _σ_1 protein forms a complex with the other chaperone glucose-regulated protein 78 kDa (GRP78/BiP). Upon ER Ca2+ depletion or by ligand stimulation, the _σ_1 protein dissociates from GRP78/BiP, leading to a prolonged Ca2+ signaling into mitochondria by IP3 receptors (Hayashi and Su, 2007). Under intracellular Ca2+ signaling disruption and subsequent ER stress, the _σ_1 protein translocates, to reach plasma membrane, recruiting Ca2+-dependent intracellular cascades (Morin-Surun et al, 1999). On the plasma membrane, it contributes to form or modify the composition of lipid-rich microdomains, so-called lipid rafts (Hayashi and Su, 2001, 2003). Increasing or activating _σ_1 proteins is expected to counteract ER stress response, whereas decreasing or inactivating them would enhance apoptosis (Hayashi and Su, 2007). Modifying _σ_1 protein activation using selective activators/agonists therefore mediates a unique pharmacological action on Ca2+ homeostasis and signal transduction pathways, which has proven to allow an effective neuroprotection against several kinds of insults, including excitotoxicity, oxidative stress, and amyloid toxicity (for reviews, see Maurice et al, 2006; Monnet and Maurice, 2006). Indeed, preliminary experiments showed that, in vitro, the selective _σ_1 activators PRE-084 and MR-22 attenuate the A_β_25–35-induced expression of the proapoptotic protein Bax and neuronal death in rat cortical cultures (Marrazzo et al, 2005). We reported that, in vivo, PRE-084 prevents the A_β_25–35-induced oxidative stress and learning impairments in mice (Meunier et al, 2006).
ANAVEX1-41 is a new aminotetrahydrofuran derivative (Vamvakides, 2002; Espallergues et al, 2007) acting as a _σ_1 protein activator, with a high affinity (44 nM) and selectivity. The CEREP profile of the compound showed that it also presents nanomolar affinities (18–114 nM) for muscarinic receptors (M1>M3, M4>M2), some low micromolar affinity for sodium channel site 2, and negligible interaction with 60 other receptor and enzyme assays (data not shown). Its molecular profile is coherent with its antiamnesic and antidepressant effects (Espallergues et al, 2007). In this study, we analyzed its antiamnesic and neuroprotective potentials against A_β_25–35-induced toxicity in mice. Learning deficits were measured using the spontaneous alternation test measuring spatial working memory and passive avoidance response measuring long-term contextual memory. The A_β_25–35-induced toxicity was also analyzed at the morphological and biochemical levels. Finally, the involvement of the _σ_1 protein or muscarinic receptors was examined using pretreatments with a selective antagonist, BD1047 or scopolamine, respectively.
MATERIALS AND METHODS
Animals
Male Swiss mice (Depré, St-Doulchard, France), aged 7 weeks and weighing 32±2 g, were used in this study. Animals were housed in plastic cages in groups. They had free access to food and water, except during behavioral experiments, and they were kept in a regulated environment (23±1°C, 40–60% humidity) under a 12 h light/dark cycle (light on at 0800 hours). Experiments were carried out between 0900 and 1700 hours, in an experimental room within the animal facility. Mice were habituated 30 min before each experiment. All animal procedures were conducted in strict adherence of European Union Directive of 24 November 1986 (86–609).
Drugs
Tetrahydro-N,_N_-dimethyl-5,5-diphenyl-3-furanmethanamine hydrochloride (ANAVEX1-41, formerly AE14) was synthesized in the laboratory (Anavex Life Sciences, Pallini, Greece). _N_-[2-(3,4-dichlorophenyl)ethyl]-N_-methyl-2-(dimethylamino)ethylamine dihydrobromide (BD1047) was from Tocris Bioscience (Bristol, UK). All other materials, including scopolamine hydrobromide, xylenol orange, and cumene peroxide, were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). Drugs used for in vivo experiments were solubilized in physiological saline solution and administered intraperitoneally (i.p.) in a volume of 100 μl per 20 g body weight. The A_β_25–35 peptide (Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met, A_β_25–35) and scrambled A_β_25–35 peptide (Ala-Lys-Ile-Gly-Asn-Ser-Ile-Gly-Leu-Met-Gly, ScA_β) were from NeoMPS (Strasbourg, France). They were dissolved in sterile bidistilled water at a concentration of 3 mg/ml and stored at −20°C until use. Before injection, peptides were aggregated by incubation at 3 mg/ml in sterile bidistilled water at 37°C for 4 days. They were administered intracerebroventricularly (i.c.v.) in a final volume of 3 μl per mouse, as previously described (Maurice et al, 1996, 1998).
Spontaneous Alternation Performances
Each mouse, naive to the apparatus, was placed at the end of one arm in a _Y_-maze (three arms, 40 cm long, 120° separate) and allowed to move freely through the maze during a single 8-min session. The series of arm entries, including possible returns into the same arm, was recorded visually. An alternation was defined as entries into all three arms on consecutive trials. The number of the total possible alternations was therefore the total number of arm entries minus two and the percentage of alternation was calculated as (actual alternations/total possible alternations) × 100. Animals performing less than eight arm entries in 8 min were discarded (ie, less than 5% of animals).
Step-Down Type Passive Avoidance Test
The apparatus consisted of a transparent acrylic cage (30 × 30 × 40 cm high) with a grid-floor, inserted in a soundproof outer box (35 × 35 × 90 cm high). A 15 W lamp lighted the cage during the experimental period. A wooden platform (4 × 4 × 4 cm) was fixed at the center of the grid-floor. Intermittent electric shocks (1 Hz, 500 ms, 40 V DC) were delivered to the grid-floor using an isolated pulse stimulator (Model 2100; AM Systems, Everett, WA, USA). The test consisted of two training sessions, at 90-min time interval, and a retention session, carried out 24 h after the first training. During training sessions, each mouse was placed on the platform. When it stepped down and placed its four paws on the grid-floor, shocks were delivered for 15 s. Step-down latency and the numbers of vocalizations and flinching reactions were measured. Shock sensitivity was evaluated by adding these two numbers. None of the treatments used in this study significantly affected the shock sensitivity. Animals that stepped down before 3 s has elapsed or that did not step down within 30 s were discarded (ie, less than 5% of the mice). Animals, which did not step down within 60 s during the second session, were considered as remembering the task and taken off, without receiving further electric shocks. The retention test was performed in a similar manner as training, except that the shocks were not applied to the grid-floor. Each mouse was again placed on the platform, and the latency was recorded, with an upper cutoff time of 300 s. Two parametric measures of retention were analyzed: the latency and the number of animals reaching the avoidance criterion, defined as correct if the latency measured during the retention session was greater than threefold the latency showed by the animal during the second training session and, at least, greater than 60 s.
Histology
Each mouse was anesthetized by intramuscular (i.m.) injection of ketamine, 80 mg/kg, and xylazine, 10 mg/kg, and quickly transcardially perfused with 50 ml of saline solution followed by 50 ml of paraformaldehyde 4%. Brains were removed and kept overnight in the fixative solution. They were cut in coronal sections (30 μm thickness) using a vibratome (Leica VT1000 S). Serial sections were selected to include the hippocampus formation and placed in gelatin-coated glass strip. Sections were stained with 0.2% cresyl violet reagent (Sigma-Aldrich), then dehydrated with graded ethanol, treated with toluene and mounted with DePeX medium (BDH Laboratories, Poole, UK). Examination of the CA1 area was performed using a light microscope (Dialux 22, Leitz), slices being digitalized through a CCD camera (Sony XC-77CE) with the NIH ImageJ software, to easily process CA1 measurement and pyramidal cells counts. Data were calculated as average of six slices and expressed as number of viable CA1 pyramidal cells per millimeter for each group.
Immunohistochemistry
Mice were anesthetized by i.m. injection of ketamine 10% and xylazine 2%, perfused transcardially with 50 ml of saline solution followed by 50 ml of paraformaldehyde 4%. Brains were removed and kept overnight in the fixative solution. Brain sections were cut in coronal sections (30 μm thickness) using a vibratome (Leica VT1000 S). Analysis of the glial response to neurodegeneration was carried out by immunolabeling sections, with mouse monoclonal antiglial fibrillary acidic protein (GFAP; Sigma-Aldrich; 1 : 1000).
Lipid Peroxidation Measures
Mice were killed by decapitation and brains were rapidly removed, weighed, and kept in liquid nitrogen until assayed. After thawing, brains were homogenized in cold methanol (1 : 10, w/v), centrifuged at 1000 g during 5 min and the supernatant collected. Homogenate was added to a solution containing FeSO4 1 mM, H2SO4 0.25 M, xylenol orange 1 mM, and incubated for 30 min at room temperature. Absorbance was measured at 580 nm (A5801), and 10 μl of cumene hydroperoxide (CHP) 1 mM was added to the sample and incubated for 30 min at room temperature, to determine the maximal oxidation level. Absorbance was measured at 580 nm (A5802). The level of lipid peroxidation was determined as CHP equivalents according to: CHP equiv.=A5801/A5802 × (CHP (nmol)) × dilution, and expressed as CHP equiv. per wet tissue weight.
Western Blotting
For determination of protein nitration levels, mice were decapitated 5 days after A_β_ peptide injection. The hippocampus were removed on ice-cold glass plate and stored at −80°C. The hippocampus tissues were homogenized in ice-cold 20 mM Tris–HCl extraction buffer, pH 7.6, containing 150 mM NaCl, 2 mM EDTA, 50 mM sodium fluoride, 1 mM sodium vanadate, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecylsulfate (SDS), 1 mg/ml pepstatin, 1 mg/ml aprotinin, and 1 mg/ml leupeptin. Equal amounts of protein, 40 μg per lane, were resolved by a 10% SDS–polyacrylamide gel electrophoresis, and then transferred electrophoretically to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). Membranes were incubated in 3% skimmed milk in a washing buffer, Tris-buffered saline containing 0.05% (v/v) Tween 20, for 2 h at room temperature. Then, membranes were incubated at 4°C overnight with an antinitrotyrosine mouse clone1A6 (Upstate Cell Signaling, Lake Placid, USA; 1 : 1000) or with goat anti _β_-actin primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA; 1 : 100). After a wash, membranes were incubated with horseradish peroxidase-conjugated antimouse IgG (Sigma-Aldrich; 1 : 2000). Peroxidase activity was revealed by using enhanced chemiluminescence (ECL) reagent. Then, intensity of peroxidase activity was semiquantified using the ImageJ software. Results were corrected with the corresponding _β_-actin level and expressed as percentage of control group data.
For determination of GFAP, caspase-3 or caspase-9 expression, mice were decapitated 7 days after A_β_ peptide injection. The hippocampi were removed on ice-cold glass plate and stored at −80°C. The hippocampus tissues were homogenized in ice-cold extraction buffer containing SDS 2% and proteases inhibitors (Roche). Equal amounts of protein, 40 μg per lane, were resolved by a 12% SDS–polyacrylamide gel electrophoresis, and then transferred electrophoretically to a nitrocellulose blot membrane (Schleicher Schuell 0.45 μm). The membranes were then blocked during 30 min at room temperature with 5% skim milk in Tris-buffered saline 20 mM (pH 7.6) containing 0.1% Tween 20 (TBS-T). The membranes were incubated at 4°C overnight with a mouse monoclonal anti-GFAP antibody (Sigma-Aldrich; 1 : 2000), or rabbit anticaspase-3 or rabbit anticaspase-9 antibody (Cell Signaling Technology; 1 : 1000 each), rinsed for 30 min in TBS-T and then incubated for 2 h with a goat antimouse or antirabbit secondary antibody (Sigma-Aldrich; 1 : 2000 each). Peroxidase activity was revealed by using ECL reagent. Then, intensity of peroxidase activity was semiquantified using the ImageJ software. Results were normalized to control values (anti _β_-tubulin; Sigma-Aldrich; 1 : 5000).
Statistical Analyses
Biochemical and behavioral data were expressed as mean±SEM, except step-down latencies expressed as median and interquartile range. They were analyzed using one-way ANOVA (F-values), followed by the Dunnett's post hoc multiple comparison test. Passive avoidance latencies were analyzed a Kruskal–Wallis nonparametric ANOVA (_H_-values), as upper cutoff times were set, followed by the Dunn's multiple comparison test. The level of statistical significance was p<0.05.
RESULTS
Antiamnesic Effects of ANAVEX1-41 Against A_β_25–35-Induced Learning Impairments
In the first series of experiments, the antiamnesic effects of ANAVEX1-41 was examined in mice centrally injected 7 days before with scrambled A_β_ (ScA_β_) or A_β_25–35 peptide. The spatial working memory was first examined in the Y_-maze test, animals receiving ANAVEX1-41 20 min before the session. As shown in Figure 1a, the central administration of ScA_β peptide or the subsequent i.p. treatment with ANAVEX1-41, in the 1–1000 μg/kg dose range, failed to change the spontaneous alternation performance that was in the 65–70% range (F<1). The treatments also did not affect the total number of arm entries (F<1; Figure 1b). When mice were treated with A_β_25–35, the alternation performance decreased highly significantly to 53% and the ANAVEX1-41 treatment reversed the deficit (F(8,145)=4.41, _p_<0.0001; Figure 1c). The compound showed a significant effect at the dose of 3 μg/kg and the improvement remained significant up to the highest dose tested. The most effective dose appeared to be 10 μg/kg. Neither the A_β_25–35, nor the ANAVEX1-41 treatments affected the number of arm entries (F(8,145)=1.62, _p_>0.05; Figure 1d).
Figure 1
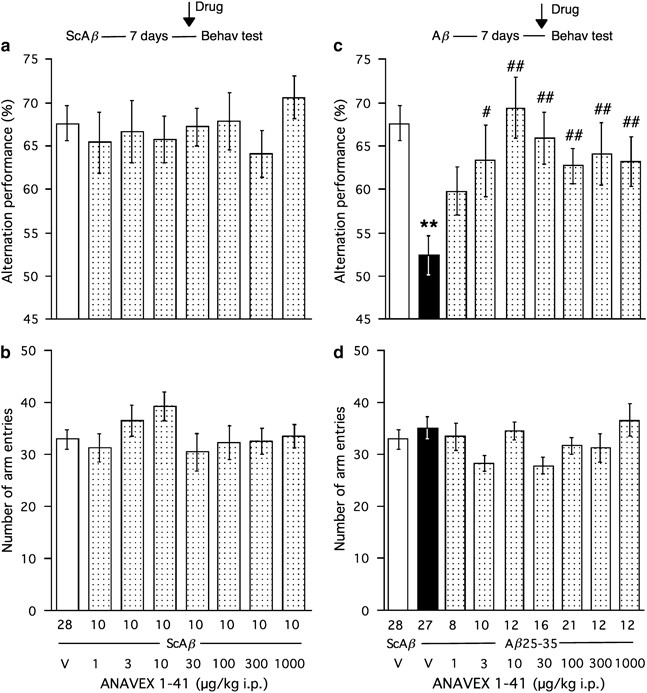
Antiamnesic effect of ANAVEX1-41 on A_β_25–35-induced spontaneous alternation deficits in mice: alternation performances (a, c) and total numbers of arm entries (b, d). Mice were injected i.c.v. with ScA_β_ or A_β_25–35 peptide (9 nmol). After 7 days, they were administered i.p. with the saline vehicle solution (V) or ANAVEX1-41 (1–1000 μg/kg), 30 min before being examined for spontaneous alternation in the Y_-maze (see insert). The number of animals per group is indicated below the columns in (b) and (d). **p<0.01 vs (Sc.A_β+V)-treated group; #p<0.05, ##p<0.01 vs (A_β_25–35+V)-treated group; Dunnett's test.
The long-term contextual memory was evaluated using the step-down type passive avoidance procedure. Animals were tested 8 days after the central administration of ScA_β_ or A_β_25–35 peptide and ANAVEX1-41 compound was administered 20 min before the first training session. The retention test was performed on day 9 after the peptide administration. As shown in Figure 2a and b, the ScA_β_ peptide or the subsequent treatment with ANAVEX1-41 in the 1–1000 μg/kg dose range, failed to affect the latency (_H_=2.98, _p_>0.05; Figure 2a) or percentage of animal-to-criterion that were in the 60–80% range (Figure 2b). In particular, the compound failed to show memory enhancing effect, as compared with V-treated animals. However, it must be noted that in procedures adapted to the measure of memory enhancing effects, the intensity of footshocks is lower than used in the present experiment. The central injection of A_β_25–35 peptide led to highly significant decreases in latency (_H_=27.72, p<0.001; Figure 2c) and percentage of animals-to-criterion (Figure 2d). The ANAVEX1-41 treatment resulted in a bell shaped but highly significant reversion of the deficits. Both parameters revealed an active dose range of 1–100 μg/kg.
Figure 2
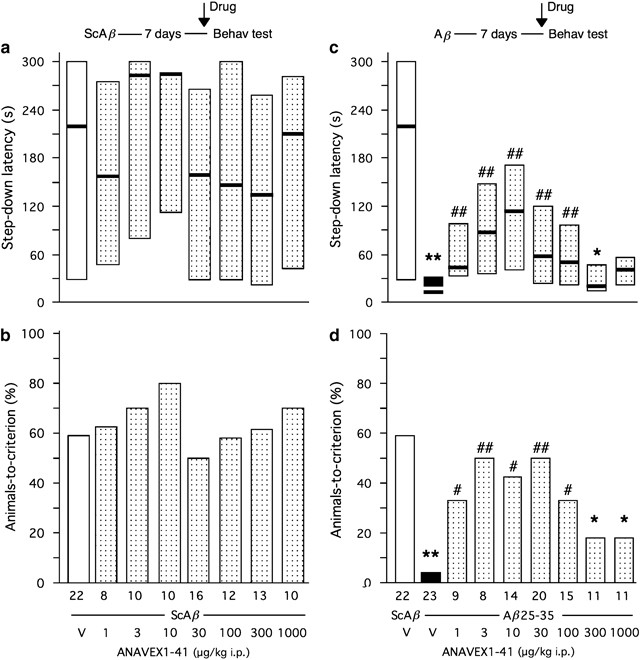
Effect of ANAVEX1-41 on A_β_25–35-induced passive avoidance deficits in mice: step-down latency (a, c) and percentage of animals-to-criterion (b, d). Mice were injected i.c.v. with ScA_β_ or A_β_25–35 peptide (9 nmol). After 7 days, they were administered i.p. with saline vehicle solution (V) or ANAVEX1-41 (1–1000 μg/kg), 30 min before the first training session (see insert). The number of animals is indicated below the columns in (b) and (d). *p<0.05, **p<0.01 vs (ScA_β_+V)-treated group; #p<0.05, ##p<0.01 vs (A_β_25–35+V)-treated group; Dunn's test in (a) and (c), _χ_2-test in (b) and (d).
Neuroprotective Effects of ANAVEX1-41 Against the A_β_25–35-Induced Learning Deficits
The neuroprotective effects of ANAVEX1-41 were first analyzed on the appearance of A_β_25–35-induced learning deficits. The drug was administered in the same, 1–1000 μg/kg i.p., dose range and only once, 20 min before the i.c.v. administration of the peptide. We previously reported that such procedure is highly effective for mixed cholinergic/_σ_1 compounds (Meunier et al, 2006). The pretreatment with ANAVEX1-41 resulted, 7 days after in a bell shaped but significant prevention of the A_β_25–35-induced spontaneous alternation impairments (F(8,145)=3.40, p<0.01; Figure 3a). The active doses of compound were in the 10–100 μg/kg range. No effect was observed in terms of number of arm entries (F(8,145)=1.64, _p_>0.05; Figure 3b). The ANAVEX1-41 pretreatment also resulted in a significant prevention of the passive avoidance deficits, both in terms of latencies (_H_=45.2, p<0.0001; Figure 3c) and number of animals-to-criterion (Figure 3d). In this procedure, however, the active dose range was 30–300 μg/kg.
Figure 3

Neuroprotective effect of ANAVEX1-41 on A_β_25–35-induced learning deficits in mice: alternation performance (a) and number of arm entries (b) in the Y_-maze test; step-down latency (c) and percentage of animals-to-criterion (d) in the passive avoidance test. Mice were administered i.p. with vehicle solution (saline, V) or ANAVEX1-41 (1–1000 μg/kg) 20 min before being injected i.c.v. with ScA_β or A_β_25–35 peptide (9 nmol). After 7 days, they were examined for spontaneous alternation or trained in the passive avoidance test (see insert). The number of animals per group is indicated below the columns in (b) and (d). *p<0.05, **p<0.01 vs (ScA_β_+V)-treated group; #p<0.05, ##p<0.01 vs (A_β_25–35+V)-treated group; Dunnett's test in (a), Dunn's test in (c), _χ_2-test in (d).
Neuroprotective Effects of ANAVEX1-41 Against A_β_25–35-Induced Toxicity
Morphological validation of the protective effect of ANAVEX1-41 was examined using the most active dose of compound, 100 μg/kg. The pyramidal cell layer of the hippocampus is highly sensitive to the amyloid toxicity observed after A_β_25–35 peptide injection in mice. We analyzed the number of viable cells in CA1 hippocampus area using cresyl violet staining (Figure 4). The A_β_25–35 injection induced a −24.6% decrease in the number of viable cells in A_β_25–35-treated mice (F(3,20)=7.68, p<0.01; Figure 4c and e) as compared with ScA_β_-treated mice (Figure 4a and e). In the same mice, no significant effect was measured in the CA3 area: 192±6 cell per field (_n_=6) for the ScA_β_ group _vs_ 187±10 cell per field (_n_=6, _p_>0.05) for the A_β_25–35 group. The ANAVEX1-41 treatment failed to affect the number of viable cells in the ScA_β_-treated group (Figure 4b and e), but significantly attenuated the diminution observed after A_β_25–35 treatment (Figure 4d and e).
Figure 4
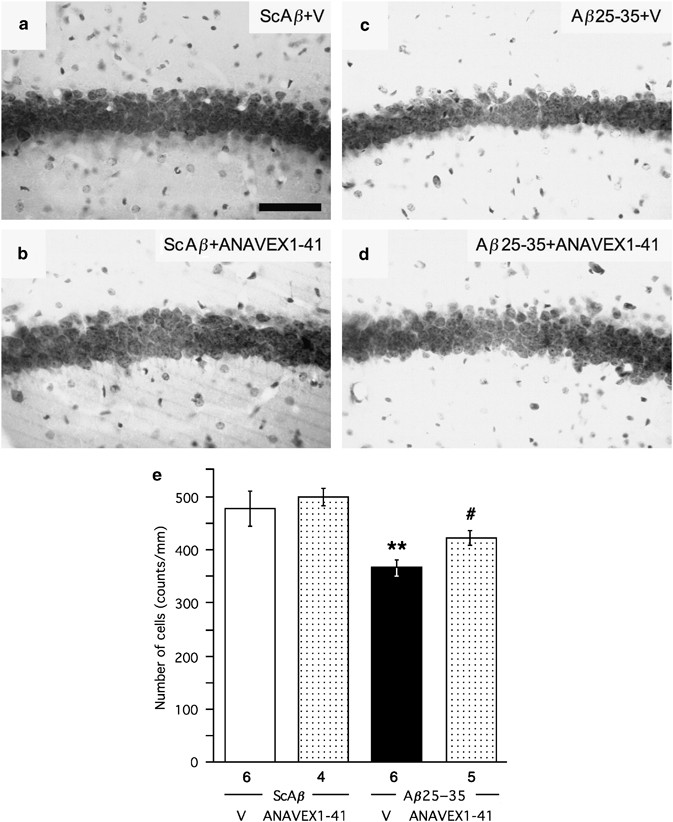
Neuroprotective effect of ANAVEX1-41 on A_β_25–35-induced toxicity in mice: pyramidal cell loss in the CA1 area of the hippocampal pyramidal cell layer, 7 days after A_β_25–35 injection. (a–d) Representative microphotographs of coronal sections of cresyl violet stained hippocampal CA1 subfield. (e) Averaged levels of viable cells. Mice were administered i.p. with saline vehicle solution (V) or ANAVEX1-41 (100 μg/kg), 20 min before being administered i.c.v. with A_β_25–35 peptide (9 nmol). Scale bar shown in (a)=100 μm. At least six slices were counted per mice and the number of mice used per group is indicated below the columns in (e). **p<0.01 vs (ScA_β_+V)-treated group; #p<0.05 vs (A_β_25–35+V)-treated group; Dunnett's test.
The extent of brain inflammation after A_β_25–35 and subsequent ANAVEX1-41 treatment was analyzed by measuring reactive astrocytes using GFAP immunohistolabeling (Figure 5). As the i.c.v. injection is expected to provoke by itself a massive glial reaction, ScA_β_-treated groups were compared with animals receiving only the i.p. treatment with vehicle solution (Figure 5a, g, m and s) or ANAVEX1-41 (100 μg/kg; Figure 5b, h, n and t). Several brain structures were analyzed and Figure 5 presents typical pictures in the retrosplenial (Figure 5a–f) and parietal (Figure 5g–l) cortices, where astrocytic clusters could be observed, and in the CA1 (Figure 5m–r) and CA3 (Figure 5s–x) areas of the hippocampus. In vehicle-treated animals, disseminated clusters containing few astrocytes were observed in the cortical areas (Figure 5a and g). The pattern of labeling was unchanged after ANAVEX1-41 i.p. injection and/or ScA_β_ i.c.v. injection (Figure 5b–d and h–j). A_β_25–35 injection, however, provoked after 7 days a marked increase in the number of labeled astrocytes and in their branching, resulting in densification of astrocytic clusters. This was observed in the retrosplenial cortex (Figure 5e), but not in the parietal area (Figure 5k). The ANAVEX1-41 treatment resulted in a blockade of A_β_25–35-induced increase of GFAP labeling (Figure 5f). In the hippocampus, astrocytes were regularly disseminated throughout the oriens and stratum radiatum layers surrounding the pyramidal cell layers (indicated by asterisks), at both the CA1 and CA3 levels (Figure 5m and s). These patterns were unchanged after ANAVEX1-41 i.p. injection and/or ScA_β_ i.c.v. injection (Figure 5n–p and t–v). The A_β_25–35 injection, however, provoked a massive densification of astrocytic labeling both in CA1 (Figure 5q) and CA3 (Figure 5w). The ANAVEX1-41 treatment resulted in an attenuation of the A_β_25–35-induced increase of GFAP labeling (Figure 5r and x).
Figure 5
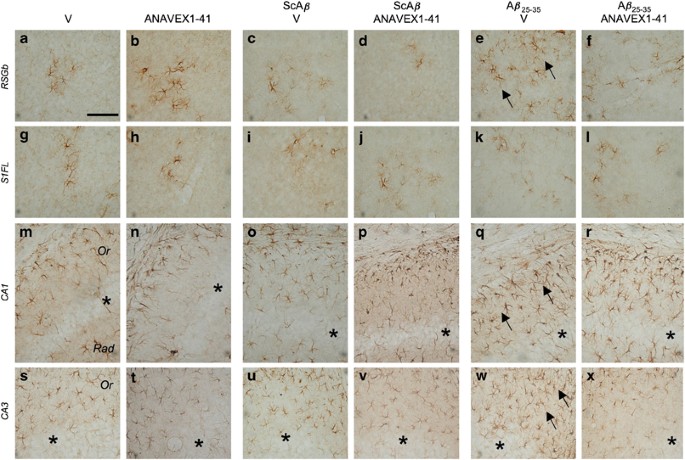
Morphological analysis of astrocytic reaction using GFAP immunolabeling in A_β_25–35-treated mice. Animals were treated i.p. with saline vehicle solution (V) or ANAVEX1-41 (100 μg/kg) and received no i.c.v. treatment (two left columns), ScA_β_ (9 nmol; two central columns) or A_β_25–35 (9 nmol; two right columns) and were killed after 7 days for immunohistological analysis. Coronal 30 μm thick sections were stained with anti-GFAP antibody and several brain areas were visually analyzed. Representative microphotographs are shown in two cortical areas, the retrospenial granular basal cortex (RSGb; a–f) and S1 cortex forelimb region (S1FL; g–l), and two hippocampal formation areas, the CA1 (m–r) and CA3 (s–x). The pyramidal cell layers are indicated by asterisks. Arrows outlined densifications of astrocyte labeling. Abbreviations: Or, oriens layer; Rad, stratum radiatum. At least three slices per mice and four mice per conditions were analyzed. Scale bar in (a)=300 μm.
Quantification of the increase in GFAP expression was performed in the hippocampus by western blotting. As shown in Figure 6, the ScA_β_ i.c.v. treatment or/and the ANAVEX1-41 i.p. treatment were without effect on GFAP expression. The A_β_25–35 treatment significantly increased GFAP expression and this increase was blocked by ANAVEX1-41 (F(5,49)=5.59, p<0.001; Figure 6). These data strengthened the qualitative immunohistochemical observations.
Figure 6
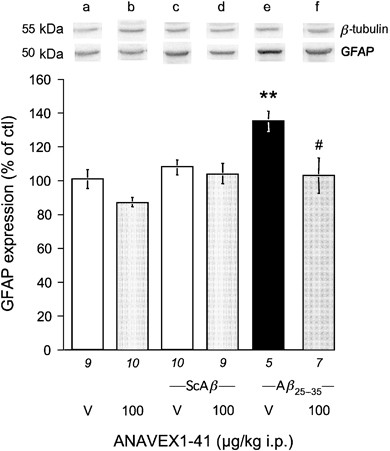
Effect of ANAVEX1-41 on GFAP expression measured by western blot in the hippocampus of A_β_25–35-treated mice. Animals were treated i.p. with saline vehicle solution (V) or ANAVEX1-41 (100 μg/kg) and received no i.c.v. treatment (two left columns), ScA_β_ (9 nmol; two central columns) or A_β_25–35 (9 nmol; two right columns) and were killed after 7 days for western blot analysis. GFAP 50 kDa variations were compared with untreated mice and normalized with β_-tubulin expression levels. Typical micrographs are shown in the upper panel. The number of animals per group is indicated below each column. The number of animals per group is indicated below the columns. Lanes on the blots: a, V; b, ANAVEX1-41; c, ScA_β+V; d, ScA_β_+ANAVEX1-41; e, A_β_25–35+V; f, A_β_25–35+ANAVEX1-41. **p<0.01 vs the V-treated group; #p<0.05 vs the (A_β_25–35+V)-treated group; Dunnett's test.
Several biochemical parameters of amyloid toxicity were also analyzed in the hippocampus extracts to validate the neuroprotective activity of ANAVEX1-41. First, amyloid peptides, and particularly A_β_25–35, induce a massive oxidative stress in forebrain structures. We therefore analyzed in the levels of lipid peroxidation (Figure 7a) and protein nitration (Figure 7b) and induction of caspase-9 expression, a marker of mitochondrial damage (Figure 7c). Second, amyloid toxicity results in cell death through caspase-dependent apoptosis pathways. We therefore measured the induction of caspase-3 expression (Figure 7d).
Figure 7
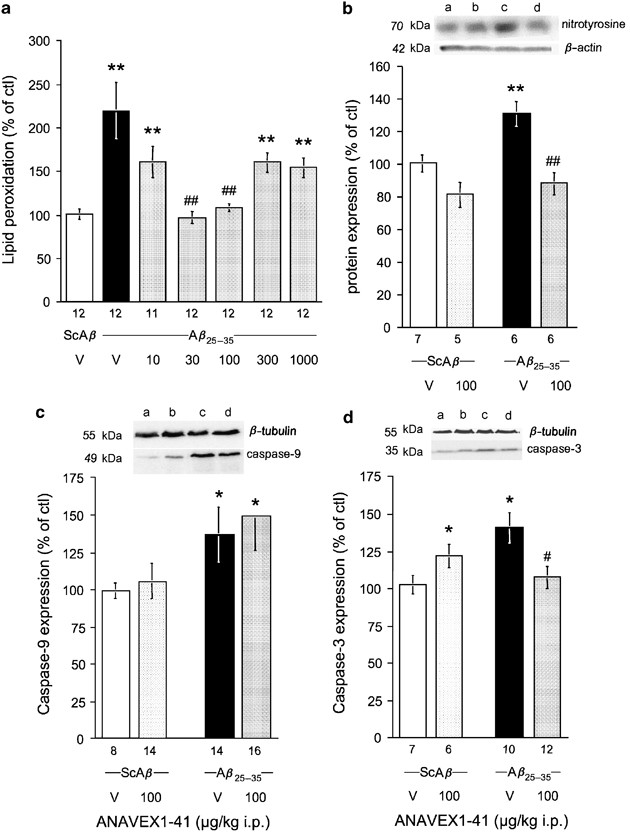
Neuroprotective effects of ANAVEX1-41 measured using biochemical markers in the hippocampus in A_β_25–35 peptide-injected mice: (a) lipid peroxidation levels, (b) protein nitration levels; (c) caspase-9 expression; (d) caspase-3 expression. Mice were administered i.p. with saline vehicle solution (V) or ANAVEX1-41, 10–1000 μg/kg in (a) or 100 μg/kg in (b) and (c), 20 min before the i.c.v. injection of ScA_β_ or A_β_25–35 peptide (9 nmol). Lipid peroxidation levels and caspases induction were measured on day 7 and protein nitration on day 5. The number of animals per group is indicated below the columns. Lanes on the blots: a, ScA_β_+V; b, ScA_β_+ANAVEX1-41; c, A_β_25–35+V; d, A_β_25–35+ANAVEX1-41. *p<0.05, **p<0.01 vs the (ScA_β_+V)-treated group; #p<0.05, ##p<0.01 vs the (A_β_25–35+V)-treated group; Dunnett's test.
A_β_25–35 induced a +117% increase in the level of peroxidized lipids that could be measured in the hippocampus (F(6,82)=8.07, p<0.0001; Figure 7a). ANAVEX1-41, tested in the 10–1000 μg/kg i.p. dose range, highly significantly, but in a U-shaped manner, prevented the A_β_25–35-induced increase in lipid peroxidation. The protective effect was measured at 30 and 100 μg/kg (Figure 7a). The western blot analysis of protein nitration revealed only a single band for nitrated proteins at the size of 70 kDa (Figure 7b, see Supplementary Figure 1 for the whole blot). A_β_25–35 induced a +30% increase in nitrotyrosine immunoreactivity (F(3,23)=8.99, _p_<0.001; Figure 7b). The pretreatment with ANAVEX1-41, 100 μg/kg i.p., tended to decrease the level of nitrotyrosine immunoreactivity in ScA_β_-treated mice (−19%, _p_>0.05) but highly significantly prevented the A_β_25–35-induced increase (Figure 7b). The western blot analysis of caspase-9 expression revealed only a single band at the size of 49 kDa that corresponded to procaspase-9 (Figure 7c). A_β_25–35 induced a +38% increase in caspase-9 expression (F(3,51)=4.13, p<0.05; Figure 7c). The pretreatment with ANAVEX1-41, 100 μg/kg i.p., failed to affect caspase-9 expression in ScA_β_- or A_β_25–35-treated animals (Figure 7c). The western blot analysis of caspase-3 expression revealed only a single band at the size of 35 kDa that corresponded to the cleaved form of caspase-3 (Figure 7d). A_β_25–35 induced a +32% increase in caspase-3 induction (F(3,34)=4.31, p<0.05; Figure 7d). The pretreatment with ANAVEX1-41, 100 μg/kg i.p., significantly prevented the A_β_25–35-induced increase (Figure 7d). However, the treatment also resulted in a significant increase in the level of caspase-3 induction in ScA_β_-treated mice (+20%, p<0.05; Figure 7d).
Involvement of (i) Muscarinic receptors and (ii) _σ_1 Protein in the Neuroprotective Effect of ANAVEX1-41
The compound is equally active, with binding affinities in the 18–114 nM range, on muscarinic M1–M4 receptors and the _σ_1 protein (Espallergues et al, 2007). To determine whether both pharmacological targets are involved in the protective effects of the compound, we coadministered: (i) the muscarinic receptor antagonist scopolamine (0.5 mg/kg) or (ii) the _σ_1 protein inactivator BD1047 (1 mg/kg) with the active doses of ANAVEX1-41 (30, 100 μg/kg). The learning abilities were analyzed after 7 days using the _Y_-maze and passive avoidance procedures. As shown in Figure 8a, the muscarinic receptor antagonist attenuated the ANAVEX1-41 effect, nonsignificantly against the 30 μg/kg dose of ANAVEX1-41 and significantly against 100 μg/kg (F(6,119)=5.14, _p_=0.0001). The BD1047 treatment led to a similar effect (Figure 8b). BD1047 attenuated the ANAVEX1-41 effect, nonsignificantly against the 30 μg/kg dose of ANAVEX1-41 and significantly against 100 μg/kg (F(6,114)=4.55, p<0.001; Figure 8b). In the passive avoidance test, scopolamine pretreatment also fully prevented the ANAVEX1-41 (100 μg/kg) effect, but not the ANAVEX1-41 (30 μg/kg) effect, similarly for latency (_H_=30.6, p<0.0001; Figure 9a) and animals-to-criterion (Figure 9b). However, different results were obtained in the contextual memory procedure with BD1047. The _σ_1 protein inactivator significantly blocked the beneficial effect of 30 μg/kg ANAVEX1-41, both in terms of step-down latency (_H_=39.7, p<0.0001; Figure 9c) and percentage of animals-to-criterion. The compound only nonsignificantly attenuated the ANAVEX1-41 (100 μg/kg) effect, particularly in terms of percentage of animals-to-criterion (Figure 9d), suggesting that protection through activation of _σ_1 protein is differentially effective on short-term and long-term memory mechanisms.
Figure 8
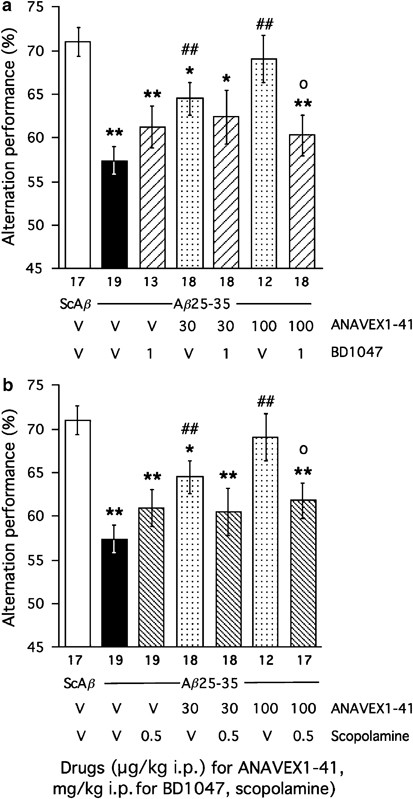
Effect of the preadministration of the muscarinic antagonist scopolamine (b) or the σ_1 receptor antagonist BD1047 (a) on the ANAVEX1-41 protective effect against the A_β_25–35-induced alternation deficits in mice. Mice were administered i.p. with saline vehicle solution (V), scopolamine (0.5 mg/kg), BD1047 (1 mg/kg) and/or ANAVEX1-41 (30, 100 μg/kg), 20 min before ScA_β or A_β_25–35 (9 nmol). After 7 days, they were examined for spontaneous alternation in the Y_-maze. The number of animals per group is indicated below the columns. *p<0.05, **p<0.01 vs (ScA_β+V)-treated group; ##p<0.01 vs (A_β_25–35+V)-treated group; °p<0.05 vs (A_β_25–35+ANAVEX1-41)-treated group; Dunnett's test.
Figure 9
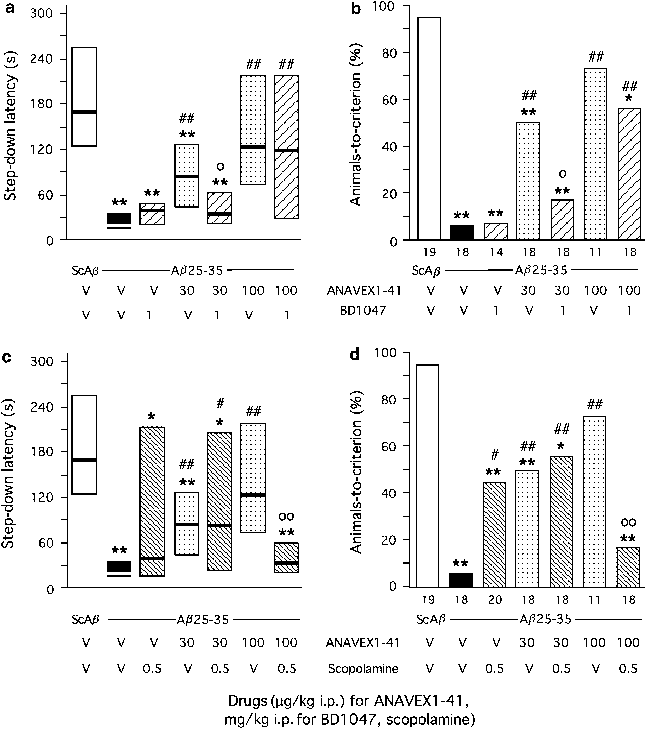
Effect of the preadministration of scopolamine (c, d) or BD1047 (a, b) on the ANAVEX1-41 effect against the A_β_25–35-induced passive avoidance deficits in mice: step-down latency (a, c) and percentage of animals-to-criterion (b, d). Mice were administered i.p. with saline vehicle solution (V), scopolamine (0.5 mg/kg), BD1047 (1 mg/kg), and/or ANAVEX1-41 (30, 100 μg/kg), 20 min before ScA_β_ or A_β_25–35 (9 nmol). They were trained on day 7 and retention was performed on day 8. The number of animals is indicated below the columns. *p<0.05, **p<0.01 vs (Sc.A_β_+V)-treated group; #p<0.05, ##p<0.01 vs (A_β_25–35+V)-treated group; °p<0.05, °°p<0.01 vs (A_β_25–35+ANAVEX1-41)-treated group; Dunn's test in (a) and (c), _χ_2-test in (b) and (d).
DISCUSSION
The first data in this study showed that ANAVEX1-41 attenuated the learning deficits observed 1 week after the central injection of A_β_25–35 peptide in mice. In the brain of rats or mice, A_β_25–35 peptide induces, after acute injection or chronic infusion, biochemical changes, morphological alterations, and behavioral impairments reminiscent of AD physiopathology. In particular, A_β_25–35-treated rodents showed spontaneous alternation, passive avoidance, and water-maze learning deficits (Maurice et al, 1996; Delobette et al, 1997) clearly related to alterations in cholinergic and glutamatergic corticolimbic systems (Maurice et al, 1996; Olariu et al, 2001). ANAVEX1-41, administered before the behavioral procedures, reversed the A_β_25–35-induced deficits with a very low active dose range because the maximum antiamnesic effect was measured at 10 μg/kg for both the short-term and long-term memory tests. This observation confirms that ANAVEX1-41 is a very potent antiamnesic drug. The compound acts as a _σ_1 protein activator, with a _K_i value of 44 nM (Espallergues et al, 2007). Such pharmacological action is known to mediate antiamnesic effects, particularly against A_β_25–35-induced learning impairments. Numerous _σ_1 protein activators including (+)-SKF-10 047, (+)-pentazocine, SA4503, or PRE-084 attenuated A_β_25–35-induced learning impairments (Maurice et al, 1998; Meunier et al, 2006). Indeed, activation of the _σ_1 protein rapidly results in amplification of Ca2+ mobilization from intracellular stores, facilitating Ca2+-dependent intracellular pathways and activation of intracellular kinases (Morin-Surun et al, 1999; Hayashi and Su, 2001; Dong et al, 2005). In turn, _σ_1 protein activators increase hippocampus glutamatergic transmission by facilitating glutamate release, activation of glutamate receptors and long-term potentiation (Monnet et al, 1992; Dong et al, 2005). They may also directly facilitate cholinergic neurotransmission by inducing acetylcholine release in the hippocampus and cortex (Matsuno et al, 1995; Horan et al, 2002).
ANAVEX1-41, however, also acts as a muscarinic ligand. We have previously reported that the compound shows _K_i values in the low nanomolar range for muscarinic receptors subtypes (18–114 nM), with a profile by ascending order of potency: M1>M3>M4>M2 (Espallergues et al, 2007). All subtypes of muscarinic receptors are expressed in the hippocampus and cortex (Levey et al, 1995) and postsynaptic M1 and autoreceptor M2 subtypes have been shown to be crucially involved in learning and memory processes (Ghelardini et al, 1999; but see also Quirion et al, 1995; Miyakawa et al, 2001; Seeger et al, 2004). Nonselective muscarinic antagonists, such as scopolamine and atropine, impair performance in various learning and memory tasks in rodents, including eight-arm radial maze learning (Eckerman et al, 1980), contextual fear conditioning (Anagnostaras et al, 1995), water-maze learning (Sutherland et al, 1982), or passive avoidance (Espallergues et al, 2007).
The combined activity of ANAVEX1-41 at _σ_1 protein and muscarinic receptors is expected to lead to synergistic effect on memory. Indeed, activation of the _σ_1 protein and M2 autoreceptors antagonism by ANAVEX1-41 (Vamvakides, 2002; Espallergues et al, 2007) may facilitate Ca2+-dependent acetylcholine release from presynaptic terminals in the hippocampus and cortex, as shown with other compounds (Quirion et al, 1995; Matsuno et al, 1995; Horan et al, 2002). As previously discussed (Espallergues et al, 2007), it is obvious that, at the very low pharmacologically active doses (10–100 μg/kg) measured for ANAVEX1-41, the compound acts both as _σ_1 activator and muscarinic receptor ligand and provokes complex concomitant effects on neurotransmission that will affect: (i) acetylcholine release, by presynaptic _σ_1 protein-mediated and M2 autoreceptor-mediated effects; (ii) phospholipase C activation induced by muscarinic receptor activation but amplified by _σ_1 protein-mediated activity; and (iii) IP3 formation and activation of ER IP3 receptors, again amplified by the _σ_1 protein activation. Noteworthy, the active dose shown by ANAVEX1-41 is unrelated to the drug in vitro affinities for either _σ_1 protein or muscarinic receptor subtypes. For comparison, PRE-084, a selective _σ_1 activator with a similar affinity of 44 nM (Su et al, 1991), is antiamnesic at 0.5–1 mg/kg against A_β_25–35-induced learning impairments (Meunier et al, 2006). One of the most promising muscarinic compound, AF102B, inhibiting 3H-quinuclidinyl benzilate binding with _K_i values in the 1–5 nM concentration range (Fisher et al, 1991), is active at 1–5 mg/kg against the learning deficits induced in rats by bilateral i.c.v. injection of the cholinotoxin ethylcholine aziridinium ion (AF64A; Nakahara et al, 1989). ANAVEX1-41, with a similar affinity for _σ_1 protein as PRE-084 and even lower affinities for muscarinic subtypes as AF102B, showed an in vivo activity at 10 μg/kg, ie, almost 100 times lower than the cited drugs. These data must be tempered after considering the protein binding and brain/plasma ratio in humans, but suggests strong synergic effects between the _σ_1 and muscarinic targets. The precise mechanism of action remains to be analyzed more adequately using in vitro preparations, but it clearly relies on facilitated Ca2+ mobilization and activation of Ca2+-dependent intracellular signaling induced by muscarinic receptor and _σ_1 protein during learning-induced neuronal activation.
The second part of the study analyzed the neuroprotective potential of ANAVEX1-41 in A_β_25–35-treated mice. For this purpose, the compound was administered at the same time as A_β_25–35, ie, 7 days before the behavioral, morphological or biochemical analyses, a procedure known to allow the observation of neuroprotective effects for mixed cholinergic and _σ_1 drugs (Meunier et al, 2006). The compound induced a bell shaped but significant prevention of A_β_25–35-induced learning deficits, with an active dose about 100 μg/kg. At the morphological level, A_β_25–35 induced a limited but significant cell loss in the CA1 pyramidal cell layer of the hippocampus (Stepanichev et al, 2004) and a marked inflammation in corticolimbic structures that could be visualized by analyzing the GFAP immunolabeling in reactive astrocytes (Stepanichev et al, 2003; Klementiev et al, 2007). Interestingly, although a significant cell loss could be measured in particularly vulnerable areas, like CA1 in mice, GFAP immunolabeling increased in a more diffuse manner, in structures associated with the amyloid deposits, as observed in the retrospenial granular basal cortex and oriens layer of the hippocampus. ANAVEX1-41, tested at 100 μg/kg, significantly attenuated the A_β_25–35-induced cell loss in CA1 and increase in GFAP expression, as shown by western blot. It appeared then that ANAVEX1-41 is able to counteract the morphological damages induced by amyloid toxicity in sensitive structures.
The neuroprotective effect of the compound was also tested using selected biochemical markers. First, A_β_ induces a strong oxidative stress, as observed in cell culture models (Behl et al, 1994) or in the hippocampus and cortex of rodents centrally injected with the peptides (Meunier et al, 2006). We therefore analyzed the level of lipid peroxidation in the hippocampus, 7 days after A_β_25–35. Peroxynitrite anion, ONOO−, is formed from nitric oxide and superoxide anion during oxidative stress and is responsible for a widespread biological damage in the AD brains (Smith et al, 1997). A_β_25–35-induced formation of ONOO− could be indirectly indicated by the level of nitrated proteins, 5 days after peptide injection (Alkam et al, 2007). Moreover, A_β_-induced oxidative stress is because of production of reactive oxygen species by the mitochondria, by premature electron leakage to oxygen through the respiratory electron transport chain, and dysfunction of enzymes responsible for limiting the superoxide production, such as NAPDH-dependent oxidase, NADH-dependent diaphorase, and superoxide dismutase (Kim et al, 2003). Several markers could be used to selectively assess the appearance of mitochondrial damage, such as release of cytochrome c into the cytosol or, as we analyzed, induction of caspase-9. Finally, we also analyzed the induction of caspase-3, known to be a key mediator of A_β_-mediated apoptosis. Results showed that ANAVEX1-41 blocked the A_β_25–35-induced increase in lipid peroxidation, at 30 and 100 μg/kg, in the hippocampus. The compound also blocked the increase in protein nitration. This antioxidant effect, however, may not primarily involve the mitochondria because A_β_25–35-induced increase in caspase-9 was not attenuated by ANAVEX1-41. Noteworthy, the _σ_1 protein is expressed at the surface of the mitochondria and at focal contacts between the ER and mitochondria (Hayashi and Su, 2007). We have previously observed that the _σ_1 protein activator PRE-084 blocks the A_β_25–35-induced increase in lipid peroxidation (Meunier et al, 2006), suggesting that activation of the _σ_1 protein results in an antioxidant effect mediated at the mitochondrial level. Our biochemical data suggest that ANAVEX1-41 also induces a strong antioxidant effect that may, however, not primarily involve a protection of mitochondrial integrity through _σ_1 protein activation. Otherwise, oxidative stress has been shown to impair M1 and M2 muscarinic receptor signaling activity, through increased phosphorylation and sequestration (Mou et al, 2006), an effect that may impede the pharmacological action of ANAVEX1-41 at muscarinic receptors. A precise mechanistic study has therefore to be carried out to identify the mechanism of the antioxidant action of ANAVEX1-41. The compound is nevertheless protective against the resulting apoptosis, as it blocked the induction of caspase-3. This observation could be considered as one of the cellular correlates of the protecting effect of ANAVEX1-41, already described at the morphological and behavioral levels.
The mechanism of the neuroprotective activity of ANAVEX1-41 is likely to involve, as detailed above regarding its antiamnesic action, a complex interaction between its muscarinic and _σ_1 targets. We observed that scopolamine or BD1047 could significantly inhibit the protective effect of ANAVEX1-41, at least in terms of learning deficits. A synergistic _σ_1/muscarinic mechanism could also be evoked to account for the neuroprotective efficacy of ANAVEX1-41, in particular, through the phospholipase C involvement and regulation of intracellular Ca2+ homeostasis.
In summary, we reported that ANAVEX1-41, a new mixed muscarinic receptor ligand and _σ_1 protein activator, is a very active antiamnesic and neuroprotective drug against A_β_25–35 peptide-induced amnesia and toxicity in the mouse. Its similar efficacy at muscarinic and _σ_1 targets suggest a unique, concomitant action, most probably at the presynaptic and intraneuronal levels, on neurotransmitter release, activation of membrane receptors and intracellular transduction systems.
References
- Abramov AY, Canevari L, Duchen MR (2004). _β_-Amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24: 565–575.
Article CAS PubMed PubMed Central Google Scholar - Alkam T, Nitta A, Mizoguchi H, Itoh A, Nabeshima T (2007). A natural scavenger of peroxynitrites, rosmarinic acid, protects against impairment of memory induced by A_β_25–35 . Behav Brain Res 180: 139–145.
Article CAS PubMed Google Scholar - Anagnostaras SG, Maren S, Fanselow MS (1995). Scopolamine selectively disrupts the acquisition of contextual fear conditioning in rats. Neurobiol Learn Mem 64: 191–194.
Article CAS PubMed Google Scholar - Behl C, Davis JB, Lesley R, Schubert D (1994). Hydrogen peroxide mediates amyloid β protein toxicity. Cell 77: 817–827.
Article CAS PubMed Google Scholar - Chafekar SM, Baas F, Scheper W (2008). Oligomer-specific A_β_ toxicity in cell models is mediated by selective uptake. Biochim Biophys Acta 1782: 523–531.
Article CAS PubMed Google Scholar - Chafekar SM, Hoozemans JJ, Zwart R, Baas F, Scheper W (2007). A_β_1–42 induces mild endoplasmic reticulum stress in an aggregation state-dependent manner. Antioxid Redox Signal 9: 2245–2254.
Article CAS PubMed Google Scholar - Delobette S, Privat A, Maurice T (1997). In vitro aggregation facilities beta-amyloid peptide-(25–35)-induced amnesia in the rat. Eur J Pharmacol 319: 1–4.
Article CAS PubMed Google Scholar - Dong Y, Fu YM, Sun JL, Zhu YH, Sun FY, Zheng P (2005). Neurosteroid enhances glutamate release in rat prelimbic cortex via activation of _α_1-adrenergic and _σ_1 receptors. Cell Mol Life Sci 62: 1003–1104.
Article CAS PubMed Google Scholar - Eckerman DA, Gordon WA, Edwards JD, MacPhail RC, Gage MI (1980). Effects of scopolamine, pentobarbital, and amphetamine on radial arm maze performance in the rat. Pharmacol Biochem Behav 12: 595–602.
Article CAS PubMed Google Scholar - Eikelenboom P, Bate C, Van Gool WA, Hoozemans JJ, Rozemuller JM, Veerhuis R et al (2002). Neuroinflammation in Alzheimer's disease and prion disease. Glia 40: 232–239.
Article CAS PubMed Google Scholar - Espallergues J, Lapalud P, Christopoulos A, Avlani VA, Sexton PM, Vamvakides A et al (2007). Involvement of the sigma1 (_σ_1) receptor in the anti-amnesic, but not antidepressant-like, effects of the aminotetrahydrofuran derivative ANAVEX1-41. Br J Pharmacol 152: 267–279.
Article CAS PubMed PubMed Central Google Scholar - Fang F, Liu GT (2006). Protective effects of compound FLZ on _β_-amyloid peptide-(25–35)-induced mouse hippocampal injury and learning and memory impairment. Acta Pharmacol Sin 27: 651–658.
Article CAS PubMed Google Scholar - Fisher A, Brandeis R, Karton I, Pittel Z, Gurwitz D, Haring R et al (1991). (+−)-_cis_-2-Methyl-spiro(1,3-oxathiolane-5,3′)quinuclidine, an M1 selective cholinergic agonist, attenuates cognitive dysfunctions in an animal model of Alzheimer's disease. J Pharmacol Exp Ther 257: 392–403.
CAS PubMed Google Scholar - Frederickson RC (1992). Astroglia in Alzheimer's disease. Neurobiol Aging 13: 239–253.
Article CAS PubMed Google Scholar - Ghelardini C, Galeotti N, Matucci R, Bellucci C, Gualtieri F, Capaccioli S et al (1999). Antisense ‘knockdowns’ of M1 receptors induce transient anterograde amnesia in mice. Neuropharmacology 38: 339–348.
Article CAS PubMed Google Scholar - Gruden MA, Davidova TB, Malisauskas M, Sewell RD, Voskresenskaya NI, Wilhelm K et al (2007). Differential neuroimmune markers to the onset of Alzheimer's disease neurodegeneration and dementia: autoantibodies to A_β_25–35 oligomers, S100 β and neurotransmitters. J Neuroimmunol 186: 181–192.
Article CAS PubMed Google Scholar - Hayashi T, Su TP (2001). Regulating ankyrin dynamics: roles of sigma-1 receptors. Proc Natl Acad Sci USA 98: 491–496.
Article CAS PubMed PubMed Central Google Scholar - Hayashi T, Su TP (2003). Sigma-1 receptors (_σ_1 binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J Pharmacol Exp Ther 306: 718–725.
Article CAS PubMed Google Scholar - Hayashi T, Su TP (2007). Sigma-1 receptor chaperones at the ER–mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131: 596–610.
Article CAS PubMed Google Scholar - Horan B, Gifford AN, Matsuno K, Mita S, Ashby Jr CR (2002). Effect of SA4503 on the electrically evoked release of 3H-acetylcholine from striatal and hippocampal rat brain slices. Synapse 46: 1–3.
Article CAS PubMed Google Scholar - Kim HC, Yamada K, Nitta A, Olariu A, Tran MH, Mizuno M et al (2003). Immunocytochemical evidence that amyloid _β_1–42 impairs endogenous antioxidant systems in vivo. Neuroscience 119: 399–419.
Article CAS PubMed Google Scholar - Klementiev B, Novikova T, Novitskaya V, Walmod PS, Dmytriyeva O, Pakkenberg B et al (2007). A neural cell adhesion molecule-derived peptide reduces neuropathological signs and cognitive impairment induced by A_β_25–35 . Neuroscience 145: 209–224.
Article CAS PubMed Google Scholar - Kubo T, Nishimura S, Kumagae Y, Kaneko I (2002). In vivo conversion of racemized _β_-amyloid ([D-Ser26]A_β_1–40) to truncated and toxic fragments ([D-Ser26]A_β_25–35/40) and fragment presence in the brains of Alzheimer's patients. J Neurosci Res 70: 474–483.
Article CAS PubMed Google Scholar - Kuboyama T, Tohda C, Komatsu K (2006). Withanoside IV and its active metabolite, sominone, attenuate A_β_25–35-induced neurodegeneration. Eur J Neurosci 23: 1417–1426.
Article PubMed Google Scholar - Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ (1995). Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci 15: 4077–4092.
Article CAS PubMed PubMed Central Google Scholar - Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, Ronsisvalle G (2005). Neuroprotective effects of sigma-1 receptor agonists against _β_-amyloid-induced toxicity. Neuroreport 16: 1223–1226.
Article CAS PubMed Google Scholar - Matsuno K, Senda T, Kobayashi T, Mita S (1995). Involvement of sigma1 receptor in (+)-_N_-allylnormetazocine-stimulated hippocampal cholinergic functions in rats. Brain Res 60: 200–206.
Article Google Scholar - Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE (1992). _β_-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12: 376–389.
Article CAS PubMed PubMed Central Google Scholar - Maurice T, Grégoire C, Espallergues J (2006). Neuro(active)steroids actions at the neuromodulatory sigma1 (_σ_1) receptor: biochemical and physiological evidences, consequences in neuroprotection. Pharmacol Biochem Behav 84: 581–597.
Article CAS PubMed Google Scholar - Maurice T, Lockhart BP, Privat A (1996). Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res 706: 181–193.
Article CAS PubMed Google Scholar - Maurice T, Su TP, Privat A (1998). Sigma1 (_σ_1) receptor agonists and neurosteroids attenuate _β_25–35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 83: 413–428.
Article CAS PubMed Google Scholar - Meunier J, Ieni J, Maurice T (2006). The anti-amnesic and neuroprotective effects of donepezil against amyloid _β_25–35 peptide-induced toxicity in mice involve an interaction with the _σ_1 receptor. Br J Pharmacol 149: 998–1012.
Article CAS PubMed PubMed Central Google Scholar - Miyakawa T, Yamada M, Duttaroy A, Wess J (2001). Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci 21: 5239–5250.
Article CAS PubMed PubMed Central Google Scholar - Monnet FP, Debonnel G, de Montigny C (1992). In vivo electrophysiological evidence for a selective modulation of _N_-methyl-D-aspartate-induced neuronal activation in rat CA3 dorsal hippocampus by sigma ligands. J Pharmacol Exp Ther 261: 123–130.
CAS PubMed Google Scholar - Monnet FP, Maurice T (2006). The sigma1 protein as a target for the non-genomic effects of neuro(active)steroids: molecular, physiological, and behavioral aspects. J Pharmacol Sci 100: 93–118.
Article CAS PubMed Google Scholar - Morin-Surun MP, Collin T, Denavit-Saubié M, Baulieu EE, Monnet FP (1999). Intracellular sigma1 receptor modulates phospholipase C and protein kinase C activities in the brainstem. Proc Natl Acad Sci USA 96: 8196–8199.
Article CAS PubMed PubMed Central Google Scholar - Mou L, Gates A, Mosser VA, Tobin A, Jackson DA (2006). Transient hypoxia induces sequestration of M1 and M2 muscarinic acetylcholine receptors. J Neurochem 96: 510–519.
Article CAS PubMed Google Scholar - Nakahara N, Iga Y, Saito Y, Mizobe F, Kawanishi G (1989). Beneficial effects of FKS-508 (AF102B), a selective M1 agonist, on the impaired working memory in AF64A-treated rats. Jpn J Pharmacol 51: 539–547.
Article CAS PubMed Google Scholar - Olariu A, Tran MH, Yamada K, Mizuno M, Hefco V, Nabeshima T (2001). Memory deficits and increased emotionality induced by _β_-amyloid25–35 are correlated with the reduced acetylcholine release and altered phorbol dibutyrate binding in the hippocampus. J Neural Transm 108: 1065–1079.
Article CAS PubMed Google Scholar - Quirion R, Wilson A, Rowe W, Aubert I, Richard J, Doods H et al (1995). Facilitation of acetylcholine release and cognitive performance by an M2-muscarinic receptor antagonist in aged memory-impaired. J Neurosci 15: 1455–1462.
Article CAS PubMed PubMed Central Google Scholar - Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J et al (2004). M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity. J Neurosci 24: 10117–10127.
Article CAS PubMed PubMed Central Google Scholar - Selkoe DJ (1989). Molecular pathology of amyloidogenic proteins and the role of vascular amyloidosis in Alzheimer's disease. Neurobiol Aging 10: 387–395.
Article CAS PubMed Google Scholar - Selkoe DJ (2004). Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 6: 1054–1061.
Article CAS PubMed Google Scholar - Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G (1997). Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci 17: 2653–2657.
Article CAS PubMed PubMed Central Google Scholar - Stepanichev MY, Moiseeva YV, Lazareva NA, Onufriev MV, Gulyaeva NV (2003). Single intracerebroventricular administration of amyloid-_β_25–35 peptide induces impairment in short-term rather than long-term memory in rats. Brain Res Bull 61: 197–205.
Article CAS PubMed Google Scholar - Stepanichev MY, Zdobnova IM, Zarubenko II, Lazareva NA, Gulyaeva NV (2006). Studies of the effects of central administration of _β_-amyloid peptide (25–35): pathomorphological changes in the hippocampus and impairment of spatial memory. Neurosci Behav Physiol 36: 101–106.
Article CAS PubMed Google Scholar - Stepanichev MY, Zdobnova IM, Zarubenko II, Moiseeva YV, Lazareva NA, Onufriev MV et al (2004). Amyloid-_β_25–35-induced memory impairments correlate with cell loss in rat hippocampus. Physiol Behav 80: 647–655.
Article CAS PubMed Google Scholar - Su TP, Wu XZ, Cone EJ, Shukla K, Gund TM, Dodge AL et al (1991). Sigma compounds derived from phencyclidine: identification of PRE-084, a new, selective sigma ligand. J Pharmacol Exp Ther 259: 543–550.
CAS PubMed Google Scholar - Sutherland RJ, Whishaw IQ, Regehr JC (1982). Cholinergic receptor blockade impairs spatial localization by use of distal cues in the rat. J Comp Physiol Psychol 96: 563–573.
Article CAS PubMed Google Scholar - Um MY, Choi WH, Aan JY, Kim SR, Ha TY (2006). Protective effect of Polygonum multiflorum Thunb on amyloid _β_-peptide 25–35 induced cognitive deficits in mice. J Ethnopharmacol 104: 144–148.
Article PubMed Google Scholar - Vamvakides A (2002). Mechanism of action of tetrahydro-N,_N_-dimethyl-5,5-diphenyl-3-furanemethanamine, a putative nootropic, anti-epileptic and antidepressant compound. Ann Pharm Fr 60: 415–422.
CAS PubMed Google Scholar - Walsh DM, Klyubin I, Fadeeva JW, Cullen WK, Anwyl R, Wolfe MS et al (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535–539.
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by collaboration contracts (no. 06122, no. 07438) between Anavex Life Sciences and INSERM (Paris, France); by grants of the ‘Academic Frontier’ Project for private universities (2007–2011) from the Ministry of Education, Culture, Sports, Sciences, and Technology of Japan; and by an exchange program between the Japanese Society for the Promotion of Science (Tokyo, Japan) and INSERM.
Author information
Authors and Affiliations
- INSERM U.710, Montpellier, France
Vanessa Villard, Julie Espallergues, Emeline Keller & Tangui Maurice - University of Montpellier 2, Montpellier, France
Vanessa Villard, Julie Espallergues, Emeline Keller & Tangui Maurice - EPHE, Paris, France
Vanessa Villard, Julie Espallergues, Emeline Keller & Tangui Maurice - Department of Neuropsychopharmacology and Hospital Pharmacy, Nagoya University Graduate School of Medicine, Nagoya, Japan
Tursun Alkam, Atsumi Nitta, Kiyofumi Yamada & Toshitaka Nabeshima - Department of Basic Medicine, College of Traditional Uighur Medicine, Hotan, China
Tursun Alkam - Department of Chemical Pharmacology, Graduate School of Pharmaceutical Science, Meijo University, Nagoya, Japan
Toshitaka Nabeshima - Anavex Life Sciences, Pallini, Greece
Alexandre Vamvakides
Authors
- Vanessa Villard
You can also search for this author inPubMed Google Scholar - Julie Espallergues
You can also search for this author inPubMed Google Scholar - Emeline Keller
You can also search for this author inPubMed Google Scholar - Tursun Alkam
You can also search for this author inPubMed Google Scholar - Atsumi Nitta
You can also search for this author inPubMed Google Scholar - Kiyofumi Yamada
You can also search for this author inPubMed Google Scholar - Toshitaka Nabeshima
You can also search for this author inPubMed Google Scholar - Alexandre Vamvakides
You can also search for this author inPubMed Google Scholar - Tangui Maurice
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toTangui Maurice.
Additional information
DISCLOSURE/CONFLICT OF INTEREST
T Maurice is a member of the scientific advisory board of Anavex Life Sciences. Other authors declare that, except for income received from their primary employer, no financial support or compensation has been received from any individual or corporate entity for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary information
Rights and permissions
About this article
Cite this article
Villard, V., Espallergues, J., Keller, E. et al. Antiamnesic and Neuroprotective Effects of the Aminotetrahydrofuran Derivative ANAVEX1-41 Against Amyloid _β_25–35-Induced Toxicity in Mice.Neuropsychopharmacol 34, 1552–1566 (2009). https://doi.org/10.1038/npp.2008.212
- Received: 25 July 2008
- Revised: 21 October 2008
- Accepted: 22 October 2008
- Published: 03 December 2008
- Issue Date: May 2009
- DOI: https://doi.org/10.1038/npp.2008.212