Targeting the Hedgehog pathway in cancer (original) (raw)
During embryonic development, the Hedgehog (Hh) signalling pathway regulates proliferation and differentiation in a time- and position-dependent fashion (for a review see Ref. 1) so that developing tissues reach their correct size with the appropriate cell types and adequate degrees of vascularization and innervation. The crucial developmental function of Hh signalling is illustrated by the dramatic consequences in human foetuses of defects in the signalling pathways, such as holoprosencephaly in Sonic Hedgehog (SHH) mutants2.
It is not difficult to imagine how a pathway that regulates both proliferation and differentiation, when mutated or otherwise deregulated, could contribute to the onset of tumorigenesis or accelerate the rate of tumour growth. Several other key signalling pathways that have crucial roles during embryonic development have been found to play a role in tumorigenesis; for example, Notch1 mutations are found in most T-cell acute lymphoblastic leukaemias (T-ALL)3, and the Wnt pathway is activated in the great majority of colon adenocarcinoma cases, often through APC (adenomatous polyposis coli) gene mutations4.
The Hh pathway has a well-recognized involvement in two types of cancer with an unmet need for drug development: basal cell carcinoma, the most common cancer in the Western world, and medulloblastoma, a childhood cancer with an invariably poor prognosis. This review provides a brief overview of Hh signalling, discusses the recent advances in the design of small-molecule and antibody-based inhibitors targeting this signalling cascade, and summarizes relatively new connections between the Hh pathway and a variety of other solid tumours.
Mechanisms of Hedgehog signal transduction
'Hedgehog' proteins (Hh) are secreted signalling proteins (Fig. 1) that were first discovered in Drosophila, along with many components of their signal transduction machinery5. They are highly hydrophobic proteins, which, after secretion, can diffuse and establish gradients in tissues, which have a paramount role in the proper development of the embryo1. In the context of embryonic development, the cells that synthesize Hh ligands are distinct from the responsive cells, which are sometimes juxtaposed to the producing cells, but can be at a significant distance.
Figure 1: A schematic of the vertebrate Hh signalling pathway in the absence or presence of Hh ligands.

The model illustrated here is based on current understanding of the Hedgehog (Hh) pathway in vertebrates. In the unliganded state (no Hh; a), Patched 1 (PTCH1), a 12-transmembrane (12-TM) domain protein, is located, at least in part, on the plasma membrane, and the 7-TM protein GPCR-like receptor Smoothened (SMOH) is predominantly located in the membrane of intracellular endosomes. It is proposed that an endogenous intracellular small molecule that acts as an agonist for SMOH (as is often true for GPCRs) is transported outside the cell by PTCH1 so that it is not able to bind to SMOH. Under these circumstances, different kinases phosphorylate GLI2/3, creating a repressor form of this transcription factor (GLIR). Iguana and SUFU prevent the active form of GLI (GLIA) from transactivating Hh-responsive genes in a manner that is still not completely understood. Upon binding an Hh ligand (b)72,73,74, PTCH1 is internalized, and apparently destabilized, so that it can no longer transport the endogenous agonist molecules outwards. This allows them to accumulate intracellularly and activate SMOH, which itself translocates to the plasma membrane, apparently concentrating in cilia in at least some types of cells. SMOH then engages as-yet-undefined components of the signalling machinery, culminating in the appearance of activator forms of GLI that then regulate the expression of Hh target genes. The known synthetic small-molecule SMOH agonists and antagonists bind to the same site as the putative endogenous ligand. Modified, with permission, from Ref. 75 © (2006) The Company of Biologists. CK1α, casein kinase 1α; GPCR, G-protein-coupled receptor; GSK3β, glycogen synthase kinase 3β PKA, protein kinase A.
Although some crucial differences exist, the signalling mechanisms are generally well conserved between Drosophila and higher organisms. Three Hh homologues with different spatial and temporal distribution patterns have been identified in humans: Sonic hedgehog (SHH), Indian hedgehog (IHH) and Desert hedgehog (DHH). The Hh signalling cascade is initiated by Hh binding to the Patched 1 protein (PTCH1 in humans, Ptch1 in mouse and Ptc in Drosophila) on the target cell (for a review see Ref. 6). In the absence of the Hh ligand, PTCH1 represses the activity of Smoothened (SMOH in humans and Smo in mouse/Drosophila), a G-protein-coupled receptor (GPCR)-like receptor, presumably by preventing its localization to the cell surface7 (Fig. 1). Binding of Hh ligand to PTCH1 causes inhibition of SMOH to be relieved and the Hh signal is transmitted via a protein complex that includes, in Drosophila, pathway-specific components such as the atypical kinesin-like protein Costal 2 (Cos2), Fused (Fu), a serine-threonine protein kinase, Suppressor of Fused (SuFu) and the zinc-finger transcription factor cubitus interruptus (Ci). Ci is regulated at several levels via phosphorylation through kinases such as protein kinase A (PKA), glycogen synthase kinase 3β (GSK3β) and casein kinase 1α (CK1α), which have a crucial role in the processing, activity and nuclear localization of Ci. In the absence of Hh signalling, Ci is cleaved, and its amino-terminal fragment acts as an inhibitor of Hh target gene transcription. Upon Hh signalling, the cleavage of Ci is prevented and the full-length molecule becomes a labile activator of Hh target gene transcription8.
However, the situation in higher organisms is not clear, because Cos2 and Fu, which are crucial for signalling in the fly, do not seem to be conserved in mammals. In contrast, mammalian Hh signalling (Fig. 1) requires the presence of non-motile cilia9,10,11 to which SMOH and other downstream pathway components need to transit in order to achieve activation of Gli transcription factors, the Ci orthologues12. Furthermore, the activator and repressor forms of Ci seem to have evolved into three separate zinc-finger proteins, with Gli1 and Gli2 functioning mostly as activators and Gli3 as a repressor13.
The mechanism by which this signalling cascade regulates proliferation is now relatively well understood and involves the activation of cyclins and cyclin-dependent kinases14. The control of differentiation might be more complex, occurring via the production of other secreted proteins, including neurotrophic and angiogenic factors15.
In general terms, it seems clear that the degree of Hh signalling in the adult is much reduced compared with that in the embryo or neonate, at least in the mouse, and has only conclusively been detected in a few sites such as CNS neural stem cells16,17,18,19 (which are responsive to Hh activation and express detectable levels of Ptch and Gli), as well as in gut epithelium20. This does not imply that the Hh pathway is without essential function in the adult, however. In addition, evidence is increasing that some of the same processes and pathways that are so prominent during development are reactivated by tissue damage and stimulate tissue repair15,21,22, including peripheral nerve regeneration23. However, in the past few years it has become clear that aberrant activation of the Hh signalling pathway can lead to cancer.
Hh pathway mutations and cancer
The first connection between aberrant Hh signalling and cancer was the discovery that the rare condition Gorlin syndrome is caused by a mutation in PTCH124,25. Gorlin patients develop numerous basal cell carcinomas (BCCs) during their lifetimes and are predisposed to other kinds of cancer as well, especially medulloblastoma, a tumour of cerebellar granule neuron progenitor cells, and rhabdomyosarcoma, a muscle tumour. More importantly, it was further determined that a large majority of sporadically occurring BCCs also involve hyper-activated Hh signalling, as judged by high levels of mRNA of the Hh target genes GLI1 and PTCH1 in tumour cells26,27. Inactivating mutations in PTCH1 occur most commonly in these tumours, with activating mutations in SMOH found in about 10% of all BCCs28,29. In addition, one-third or more of all human medulloblastoma cases have been shown to involve increased Hh signalling, often due to PTCH1 and sometimes to SUFU mutations30. In all of these cases, it is believed that deregulated Hh signalling leads to increased cell proliferation and tumour formation.
Experiments in various mouse models have generally confirmed these observations. Overexpression of Shh31, Gli132, Gli233 or constitutively active Smo29 in the skin of otherwise normal mice produces lesions that are BCC-like in histological appearance and in expression of various phenotypic markers. Mice that are heterozygous for a Ptch1 mutation (that is, a mouse model of Gorlin syndrome) are larger than normal, have a higher frequency of spontaneously occurring medulloblastoma and are more susceptible to UV-induced BCC, much like Gorlin patients34. Finally, mice carrying heterozygous loss-of-function mutations in SuFu also develop a skin phenotype resembling the one seen in Gorlin syndrome35.
These results implicate the Hh pathway as an important pharmacological target for a variety of conditions. Hh agonists currently in preclinical development for neuroprotection/regeneration36 will not be discussed here. Hh antagonists have also shown promising results, particularly for the treatment of BCC, medulloblastoma and a variety of solid tumours in preclinical models.
Effects of Hh antagonists in models of BCC
With almost 1,000,000 new occurrences per year in the US alone, BCC is the most common cancer in the Western world. The clear connection between activation of Hh signalling in basal cells of the skin and the formation of BCC lesions led to a series of studies designed to identify small-molecule Hh antagonists37,38,39. As the Hh pathway in BCC is affected at the level of PTCH1 or SMOH, such antagonists must act at, or downstream of, SMOH in the Hh signalling cascade to be of therapeutic value. The most widely used and commercially available inhibitor of the Hh pathway is cyclopamine, a natural-product SMOH inhibitor derived from corn lilies. A few groups have tried to identify other Hh antagonists, especially those that are completely synthetic. Chen et al.39, using a cell-based screen formatted around a luciferase reporter under the control of a Gli1-binding site, identified a few chemically distinct classes of antagonist, all of which seemingly bound to SMOH, albeit potentially at different sites (Table 1). Williams et al. reported on a similar cell-based assay that eventually yielded several different classes of Hh antagonists, some of which have been further optimized by a significant amount medicinal chemistry38. One such class, represented by the aminoproline Cur-61414 (Curis), directly binds to SMOH and effectively inhibits the Hh pathway37,38. This compound prevented the formation of BCC-like 'basaloid nests' in Shh-treated ex vivo skin punches from Ptch1 +/− embryonic mice and, more importantly, also eliminated preformed BCC-like lesions. The lesions disappeared as a result of decreased proliferation and increased apoptosis, seemingly with no effect on the surrounding normal skin. Cur-61414 also selectively induced apoptosis and eliminated BCC-like lesions induced in UV-irradiated skin explants of Ptch1 +/− mice. Furthermore, prolonged systemic treatment with cyclopamine significantly diminished tumour formation in UV-irradiated Ptch1 +/− mice, with no obvious deleterious effects40.
Table 1 Summary of selective Hedgehog pathway inhibitors
These in vitro and in vivo results suggested that inhibiting the Hh pathway could block tumour growth and stimulate tumour regression without any obvious toxic effects on normal adjacent tissue. Cur-61414 was therefore formulated as a topical agent, which seemed to be safe and well tolerated in preclinical models41, and is the first class of Hh antagonist to have entered a formal clinical trial, having been tested in a Phase I study with BCC patients42. A small 'physician-sponsored' study found topical treatment with cyclopamine to be effective in reducing the size of BCC lesions43.
Effects of Hh antagonists on medulloblastoma
Medulloblastoma is the other cancer with a well-recognized involvement of the Hh pathway. Although fewer than 500 children per year in the US are diagnosed with medulloblastoma, the outcome is almost invariably poor. Surgery with subsequent radiation or chemotherapy has increased survival to greater than 50%, but there is severe treatment-associated morbidity, including mental retardation. Clearly, a therapeutic regimen that eliminates tumour cells more selectively would provide a great improvement over existing therapies.
During normal development, Shh is a mitogen for cerebellar granule neuron progenitors44,45,46 and constitutive activation of this pathway in these cells seems to give rise to medulloblastoma. Therefore, Hh-pathway antagonists have been tested in cell culture and mouse models of medulloblastoma. Cyclopamine can decrease the rate of growth of mouse medulloblastoma cells both in culture and in mouse allograft models47,48. Scientists at Curis and Genentech identified and extensively chemically modified a new class of SMOH-binding Hh antagonists that proved to be very potent, with excellent oral bioavailability. Without intervention, 90% of Ptch1 +/−, p53 −/− mice spontaneously develop medulloblastoma, but if treated with one of these new Hh antagonists the tumours could be completely eliminated, again by the dual activity of blocking tumour-cell proliferation and stimulating apoptosis, leaving behind a normal looking cerebellum49. Effects in this model were also demonstrated with cyclopamine50. Further work will be needed to assess cerebellar function (and other aspects of CNS function) subsequent to tumour elimination.
Surprisingly, in vitro medulloblastoma experiments have been more challenging. Proliferation of newly dissociated mouse primary cerebellar granule neuron progenitor cells (which give rise to medulloblastoma) was sensitive to Hh antagonists at concentrations similar to those needed to inhibit Hh signalling49. However, following serial passaging of medulloblastoma cells in vitro, much higher concentrations of Hh antagonists were required to block proliferation than to block Hh signalling49,51. In other words, it seems that Hh signalling becomes less important in controlling the rate of proliferation as the cells are passaged in vitro, and that the high concentrations of Hh antagonists needed to inhibit proliferation might reflect non-specific or toxic effects of these compounds.
One of the more interesting in vitro observations relates to the effects of cyclopamine on growth of primary human medulloblastoma cells cultured immediately in vitro48. Growth of each of seven independent sets of cells was inhibited by cyclopamine, suggesting that Hh inhibition might have a more general effect on these cells than anticipated, as less than one-third of the tumours would be predicted to have Hh-pathway mutations. Hh antagonists might have the capacity to block the growth of all medulloblastomas, presumably because Hh has a mitogenic effect on these cells even when the pathway is not hyperactivated through mutation. If confirmed, these results could support the notion that Hh antagonists could be broadly effective in treating this type of brain tumour.
Hh ligand overexpression in solid tumours
There is little convincing evidence connecting activating mutations of the Hh pathway to tumour types other than BCC and medulloblastoma. However, given that pathways controlling cell proliferation are implicated in cancer development, it was of interest to identify cancers involving excessive Hh signalling. Hh affects the development and repair of certain tissues such as lung52,53, bone54,55 and pancreas56, and so it can be proposed that Hh-dependent cancers might arise from such tissues. In addition, it had been previously speculated that Hh ligand expression might exert growth-promoting effects in certain cancers26.
Some of the predictions based on these types of known effects of Hh have been difficult to validate. For example, although Shh exerts a well-known influence on CNS development, convincing evidence for its role in brain tumours has been restricted to medulloblastoma, although recent experiments have implicated Hh signalling in glioma47. A similar situation is found with respect to cartilage and bone. Ihh has a clear mitogenic effect on prehypertrophic chondrocytes, and bone formation is reduced considerably in Ihh −/− mice55, yet only a few studies find excessive Hh signalling in bone/cartilage tumours such as endochondroma, osteochondroma and chondrosarcoma57,58.
However, characterization of human tumour biopsies and tumour cell lines for expression of Hh ligands and downstream pathway components such as PTCH1, SMOH and GLI followed by treating tumour cell lines in vitro with high cyclopamine concentrations, and use of standard mouse xenograft models, have pointed to the Hh pathway as having a truly central role in the growth of a vast array of cancer types. The first identified Hh-overexpressing tumour was small-cell lung cancer (SCLC)59, studied in part because Hh is known to be essential for branching morphogenesis in the developing lung53. Watkins et al. first demonstrated that naphthalene-induced damage to murine airway epithelial cells stimulated reactivation of Hh signalling by upregulating production of Shh and Gli1, both, unexpectedly, within epithelial cells (as opposed to the more common mode of signalling in which Ptch1/Gli1 levels would be expected to be high in the stromal compartment)59. They further discovered that about 25% of human SCLC samples had relatively high expression of both SHH and GLI1 and that growth of SCLC cell lines could be blocked in vitro using cyclopamine or a monoclonal anti-Hh antibody (5E1) that prevents Hh from binding to PTCH1. Furthermore, systemic administration of cyclopamine was effective in a nude mouse SCLC xenograft model59. These results were particularly important in raising the prospect of tumour types whose growth was regulated in an autocrine or juxtacrine fashion by tumour cell-derived over-production of Hh ligand.
Of the many types of cancer in which the Hh pathway has subsequently been implicated, special attention has been paid to pancreatic cancer, other tumours of the gastrointestinal (GI) tract and prostate cancer. Thayer et al.60 showed abnormal expression of SHH, PTCH1 and SMOH early in the formation of human pancreatic tumours. PTCH1 was found in both epithelial and stromal compartments, providing the potential for autocrine and paracrine signalling. Several pancreatic cancer cell lines were found to be PTCH1- and SMOH-positive and growth-inhibited by cyclopamine in vitro, suggesting an active autocrine loop through which tumour cells both make and respond to Hh ligand. More importantly, the growth of these cell lines xenografted into nude mice was also slowed by systemic treatment with cyclopamine. Similar observations were made by Berman and colleagues61 for pancreatic and other GI tumours, although they emphasized the autocrine activity of Hh ligand in that the growth of some of the cell lines could be accelerated by added Hh ligand and blocked either by cyclopamine or by 5E1, both in vitro and in vivo. Several groups have now provided similar data for prostate cancer as well, including SHH overexpression in tumour biopsies (particularly in higher gleason grade tumours) and in vitro and in vivo inhibitory effects of cyclopamine on growth of prostate cancer cell lines62,63,64. Strikingly, Karhadkar et al.62 implicated the Hh pathway in prostate tumour metastasis, as the capacity of AT6.3 cells to metastasize to the lung was completely abrogated by cyclopamine, and a rarely metastasizing clone, AT2.1, could be induced to metastasize by overexpression of Gli1, in a cyclopamine-insensitive manner. This could be explained if Hh exerts its effect through the induction of an epithelial–mesenchymal transition via the upregulation of the transcription factor SNAIL and the downregulation of E-cadherin. Consistent with a direct effect on the metastatic process, cyclopamine was shown to inhibit cell migration through a Matrigel matrix in vitro62.
Although most of these studies report an autocrine requirement for the pathway in tumour cells, it should be remembered that the activities of Hh during development are typically paracrine, raising the confounding issue that the proliferation of certain types of tumour cells might be blocked with Hh antagonists in vivo (where tumour cells can signal to surrounding stromal cells and vice versa), even when it is not in vitro (where stromal cells are generally absent). For example, during development, Shh produced by the prostatic epithelium signals to the underlying mesenchyme, and it remains somewhat controversial whether Hh ligand produced by tumour cells signals to the tumour stroma and induces the production of growth factors supporting tumour growth/survival65 or if the tumour switches to an autocrine requirement for Hh signalling where the tumour cells both produce and respond to the ligand62,63,64 (Fig. 2). It could be that, in some cases, both paracrine and autocrine mechanisms are involved so that Hh overexpression by the tumour cells orchestrates effective tumour growth by direct stimulation of tumour-cell proliferation in an environment rich in supporting survival and angiogenic factors. In these cases, the effects of Hh overexpression might be particularly deleterious clinically, suggesting that Hh antagonists might also be particularly effective. This adds another layer of complexity to efforts designed to ascribe tumour growth to excess Hh activity. Figure 3 provides an overview of all types of cancer in which the Hh pathway has been implicated.
Figure 2: Models of Hh pathway activation in cancer.

a | Type 1: loss-of-function mutations in Patched 1 (PTCH1; yellow star) or gain-of-function mutations in Smoothened (SMOH; blue star) lead to constitutive Hedgehog (Hh) pathway activation. b | Type 2: autocrine model in which tumour cells produce and respond to Hh ligand. Pathway activation may occur in all tumour cells or in a small number of tumour stem cells. c | Type 3: paracrine model in which tumour cells produce Hh ligand and surrounding stromal cells respond by producing additional growth factors to support tumour growth or survival, for example. IGF, insulin-like growth factor; VEGF, vascular endothelial growth factor.
Figure 3: Types of cancer with possible Hedgehog deregulation.
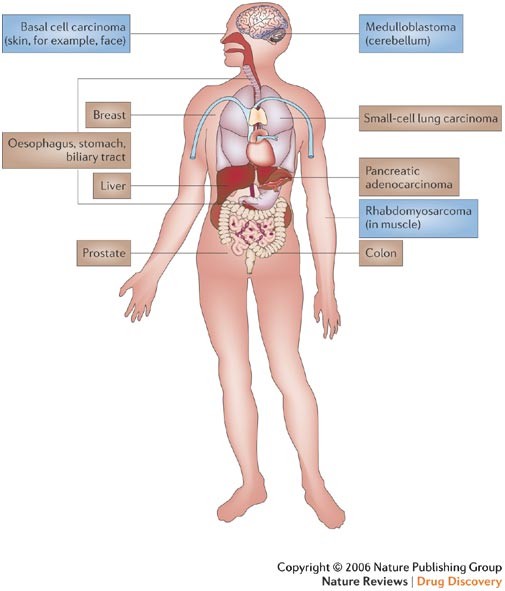
Mutations of Patched 1 (PTCH1) and Smoothened (SMOH) have been found in basal cell carcinoma24,25,29,77 and mutations in PTCH1 and Suppressor of Fused (SUFU) have been found in meduloblastoma77,78 and rhabdomyosarcoma79. An autocrine requirement of Hedgehog (Hh) ligand has been reported for small-cell lung carcinoma59, pancreatic adenocarcinoma60,61, oesophageal, stomach and biliary tract cancers61, prostate cancer62,63,64,65, breast cancer80, colon cancer81,82,83 and liver cancer84. Type 1 cancers (with mutations in the Hh pathway) are in blue boxes, type 2 cancers (autocrine requirement for Hh ligand) are in brown boxes.
Hh signalling and cancer stem cells
The theory that many tumours arise from a small number of cancer stem cells has recently gained strength with some landmark studies that point to the existence of a discrete population of slowly dividing tumour cells capable of self-renewal and differentiation along multiple lineages66. The control of these processes in cancer stem cells is thought to be regulated by a small number of signalling pathways such as Hh, Wnt, Notch and BMP, and growing evidence suggests that some of these pathways are deregulated, allowing for their abnormal expansion and the formation of cancer. One of the best-characterized model systems to date is that of the mammary gland, where in vitro cultures and reconstitution experiments in non-obese diabetic (NOD)/severe combined immunodeficiency (SCID) mice have identified such a stem-cell population both in normal breast tissue and in breast tumours. Liu et al.67 have shown that overexpression of Wnt ligand or of constitutively active β-catenin in mammary gland leads to an increase in mammary stem-cell numbers. Similarly, Hh stimulation in culture increased the number and size of mammary stem-cell aggregates (referred to as mammospheres), whereas cyclopamine had the opposite effect68,69. Finally, the Notch pathway also seems to have a role in mammary stem cells as shown using γ-secretase inhibitors or activated DSL peptide70. These three pathways might be interconnected and ultimately lead to the regulation of stem-cell self-renewal via the Bmi1 (B lymphoma Mo-MLV insertion region 1) transcription factor69. These data suggest additional possible uses for inhibitors of pathways such as Hh in the treatment of cancer — that is, directly targeting the proliferation of the cells that ultimately give rise to the tumours.
Conclusions
The increased understanding of the role of morphogenic pathways — particularly the Hh pathway — in development and cancer provides a new and exciting opportunity for therapeutic intervention. First, cancers such as medulloblastoma and BCC, in which mutations lead to constitutive pathway activation, should be the most sensitive to pathway inhibition and might provide the fastest and most straightforward path to developing therapeutics targeting SMOH or other components downstream of the mutated proteins. The recent identification of tumours requiring Hh ligand-mediated activation of the pathway could provide a much larger opportunity where therapeutic effects might be achieved by Hh pathway inhibition, not only with the same small molecules used to target tumours where the pathway is mutated, but also with blocking antibodies or other soluble receptor fragments directed at the ligand(s) themselves.
However, a significant number of challenges still need to be overcome for the successful clinical development of an Hh-pathway inhibitor. Although the current literature seems to indicate that inhibition of the Hh pathway is relatively safe in adults, appropriate toxicology studies need to be performed. This will be particularly important for treating medulloblastoma, where the use of Hh-pathway antagonists will be mostly directed at children in whom the pathway might still be active in normal tissues. In addition, there is recent evidence indicating that the Hh pathway might be important for regulating stem cells in various tissues, and particular attention will be needed to ascertain the potential effects on these stem cells as well as on tissue regeneration and tissue repair.
It is thought that Hh- or Wnt-driven pathway activities could be important in up to one-third of cancers71. However, the exact contribution of Hh in these tumours remains to be clarified, because the Hh signal can potentially contribute to the proliferation and/or survival of tumour cells or of a more discreet tumour stem-cell population. Alternatively, or in addition to its autocrine role, tumour-derived Hh ligands might act in a paracrine mode to stimulate a stromal or angiogenic response. Better understanding of the role of Hh in these tumours will help to identify good predictive and pharmacodynamic markers of tumour responses, which will be essential for the successful development of Hh antagonists. Although the challenge remains large, it will be exciting to explore the outcome of treating various cancers with such Hh-pathway inhibitors.