Dysosteosclerosis is also caused by TNFRSF11A mutation (original) (raw)
Introduction
Dysosteosclerosis (DOS: MIM %224300) is rare sclerosing bone disease which belongs to the group 23 of genetic skeletal disorders (‘Osteopetrosis and related disorders’) [1]. DOS is characterized by osteosclerosis and platyspondyly, and is often complicated by short stature, fractures, cranial nerve palsy, poorly calcified enamel, skin changes such as macular atrophy and flattened fingernails [2,3,4]. The radiological manifestations of DOS consist of sclerotic skull, flattened and diffusely dense vertebral bodies, expanded metaphyses of the long tubular bones with irregular sclerosis [2,3,4]. The transmission in reported families supports its autosomal recessive inheritance [2,3,4].
Campeau et al. [5] identified homozygous and compound heterozygous missense mutations of solute carrier family 29, member 3 (SLC29A3, MIM 612373) in two unrelated patients with DOS. SLC29A3 encodes a nucleoside transporter, which is expressed in osteoclasts and essential for lysosomal function [6]. No other SLC29A3 mutation in DOS has been published to our knowledge. There are many cases of DOS without SLC29A3 and hence its genetic heterogeneity has been suggested.
In this study, we reported a Turkish patient with DOS. By whole-exome sequencing of the patient, we identified a homozygous splice-site mutation in TNFRSF11A coding tumor necrosis factor receptor superfamily member 11a (alias: receptor activator of nuclear factor-kappa B, RANK) that caused skipping of an exon.
Materials and methods
Patient
A 16-years-old Turkish girl was referred to us because of recurrent fractures and bowing of femurs. She is the youngest child of healthy consanguineous parents who have three healthy girls, one healthy boy and two abortions (Figure S1). The patient was born as a term baby with birth weight 3010 g. Prenatal and postnatal course were uneventful until age three years when she was suspected for osteogenesis imperfecta during treatment of pneumonia elsewhere. She had no fracture until age 15 years when she fell from her bike and had right femoral fracture, which needed operation. At age 16, she had a left femoral fracture after falling down and was admitted to our hospital.
On examination, her height was 150 cm (<3rd percentile), weight 45 kg (<3rd percentile) and occipito-frontal circumference 52.5 cm (~25 percentile). She had long shape face, mild proptotic eyes with white sclera, long and wide nose, hypoplastic uvula, and irregular teeth. She had no evidence for easily bruisability and cranial nerve palsy. She had no specific changes on the skin such as macular atrophy and flattened fingernails. There was no history of recurrent infection or susceptibility to infection. Laboratory examinations including blood count and serum immunoglobin levels were unremarkable.
A skeletal survey showed diffuse osteosclerosis of craniofacial bones, which was most prominent in the skull base (Fig. 1a, b). Endplates and neural arches of the spine were sclerotic and vertebral bodies, particularly its posterior half were flattened; shafts of ribs and bodies of claviculae and scapulae showed sclerosis, but the costal and clavicular ends and glenoids were radiolucent (Fig. 1c, d). Pelvic bones and proximal femora showed sclerosis, which were most prominent in femoral shafts and iliac bodies (Fig. 1e). The long bones showed only mild diaphyseal sclerosis and metaphyseal radiolucency with severe metaphyseal under-modeling (Erlenmeyer flask deformity) (Fig. 1c, f). The short tubular bones were normal (Fig. 1g). There was apparent bone fragility, including a transverse fracture in the left femoral shaft, old fracture in the right femoral shaft, and old callus in the fibular shaft. Thus, the skeletal phenotype of the patient was quite compatible to DOS. DEXA of the spine (L1–L4) showed marked increase of bone mineral density (BMD: 2.03-2.54 g/cm2, Z score 6.9–9.7).
Fig. 1
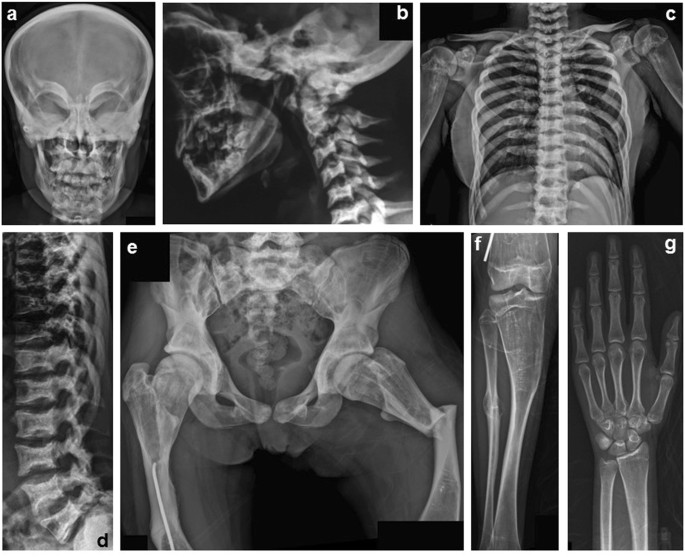
A skeletal survey of the patient with osteosclerosis at age 16 years. a, b Craniofacial bones show diffuse osteosclerosis, which is prominent in the skull base. The cervical spine is also sclerotic. c, d Ribs and scapulae show sclerosis, but the costal ends and glenoids are radiolucent. Clavicular shafts are sclerotic, whereas clavicular ends are radiolucent. The proximal clavicular ends are expanded. The proximal humeri are under-modeled with patchy sclerotic foci in metaphyses. Vertebral endplates and neural arches of the thoracolumbar spine are sclerotic. The vertebral bodies, particularly its posterior half are flattened. e Pelvic bones and proximal femora show sclerosis, which are most prominent in femoral shafts and iliac bodies. There is a transverse fracture in the left femoral shaft. An old fracture in the right femoral shaft is surgically treated. f Femur, tibia, and fibula show prominent metaphyseal under-modeling (Erlenmeyer flask deformity). Their metaphyses are radiolucent, while their diaphyses are mildly sclerotic. Note callus in the fibular shaft. g Distal radial end shows minimum under-modeling. Osteosclerosis is absent in wrist and hand
Whole-exome sequencing and variant calling
The study protocol was approved by the ethical committee of RIKEN [approval number: H16-40 (12)] and participating institutions. Peripheral blood was obtained from the family members after the informed consent. Genomic DNA was extracted from the blood using QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). DNA concentration was measured by using a Qubit V.2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).
Whole-exome sequencing was performed as previously described [7,8,9,10,11]. Briefly, DNA (3 μg) was sheared with S2 Focused-ultrasonicator (Covaris, Wobum, MA, USA) and processed by SureSelectXT Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA). Captured DNA was sequenced using HiSeq 2000 (Illumina, San Diego, CA, USA) with 101 bp pair-end reads with seven indices. Image analysis and base calling were performed using HCS, RTA, and CASAVA software (Illumina). Reads were mapped to the reference human genome (hg19) by Novoalign-3.02.04. Aligned reads were processed by Picard to remove PCR duplicates. Variants were called by GATK v2.7-4 following GATK Best Practice Workflow v3 [12] and annotated by ANNOVAR [13].
PCR and Sanger sequencing
The mutation identified by the whole-exome sequencing was confirmed by Sanger sequencing. The region of genome including the mutations was amplified by PCR and sequenced both strands. The primers were 5′-CCCACAGCTGTACCTTCCTT-3′ and 5′-CATGCACGGGATGAAATAAA-3′. A 3730 DNA analyzer (Life Technologies) was used for the Sanger sequencing. Sequencher V.4.7 (Gene Codes, Ann Arbor, MI, USA) and Genetyx (Genetyx, Tokyo, Japan) were used for aligning sequencing chromatographs to reference sequences.
Evaluation of the mutation identified in TNFRSF11A
The variants identified by the exome sequencing were evaluated by using five databases, dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (http://www.1000genomes.org/), ExAC (http://exac.broadinstitute.org/), ESP6500 (http://evs.gs.washington.edu/EVS/), and Human Gene Mutation Database (HGMD: https://portal.biobase-international.com/hgmd/pro/start.php). Homozygosity mapping was performed by Homozygosity Mapper as previously described [14] on the whole-exome sequencing data. We also evaluated the pathogenicity of the variant using several online splice-site prediction programs, such as Neural Network Splice Site Prediction (NNSPLICE: http://www.fruitfly.org/seq_tools/splice.html), SplicePort (http://spliceport.cbcb.umd.edu/), Alternative Splice Site Predictor (ASSP: http://wangcomputing.com/assp/), Human Splice Finder (HSF: http://www.umd.be/HSF/), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html).
Exon trapping assay
Exon trapping assay was performed as previously described [15, 16]. Briefly, a 3956-bp genomic region encompassing exons 5–7, full length of intron 5 and 6, partial intron 4 and 7 of TNFRSF11A (Fig. 2b) in the patient and her elder sister was amplified by PCR using the primer: 5′-ACCCCTCGAGCAAGCCTGTGGGAGCTGAAG-3′ and 5′-CCGCTCTAGACCTGAACGCCAGCGTATTCC-3′. PCR products were digested with _Xho_I and _Xba_I, and cloned into an exon trapping vector (Mo Bi Tec, Goettingen, Germany). Exon trapping vectors were transfected into COS-7 and Hela cells, respectively. Total RNAs were isolated using SV Total RNA Isolation System (Promega, Madison, WI, USA). RT-PCR was performed using primer sets supplied with the exon trapping assay kit and PCR products were sequenced.
Fig. 2
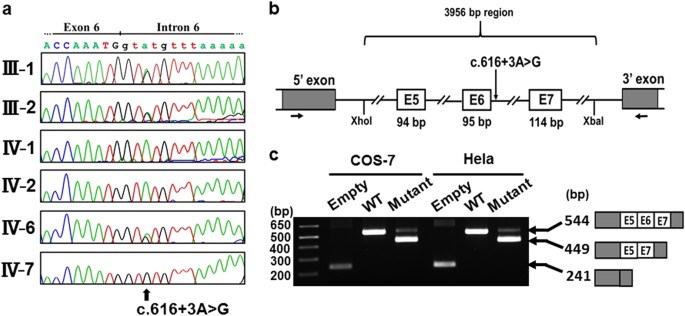
TNFRSF11A mutation. a Electropherograms of Sanger sequence of the family. A homozygous mutation (c.616+3A>G) is found in the patient (IV-7); the parents (III-1 and III-2) and a normal sibling (IV-6) are heterozygous for the mutation. b, c Exon trapping assay. b The patient’s TNFRSF11A genomic sequence spanning exons 5–7, full length of intron 5 and 6, partial intron 4 and 7 corresponding to the wild type and mutant sequences, was cloned into Exontrap vectors. c The reverse transcriptase polymerase chain reaction (RT-PCR) following transfection into COS-7 and HeLa cells. Note that the wild type vector trapped exons 5–7 but the mutant vector caused skipping of exon 6
Results and discussion
We performed the whole-exome sequencing in the patient and harvested about 2.5 Gb sequences. The sequences were successfully mapped to all human RefSeq. At least 97.2% of all coding regions were covered in a depth of 10 reads (Table S1). We identified a homozygous splice-site variant (NM_003839.3: c.616+3A>G) in TNFRSF11A encoding receptor activator of NF-kappa-B (RANK). The variant was not deposited in any available databases, including dbSNP, 1000 genomes, ExAC, ESP6500, and HGMD. Homozygosity mapping using the whole-exome sequencing data showed that TNFRSF11A is residing in an 18.6 Mb homozygous stretch of the patient genome (Table S2). By Sanger sequencing, we confirmed the variant in the patient. The variants were heterozygous in her parents and a normal sibling (Fig. 2a). Likely disease causing variants in other known causal genes of osteosclerosis including SLC29A3 were not identified in the dataset.
c.616+3A>G is located in the splice donor site of intron 6 of TNFRSF11A. Several in silico prediction tools for splice-site variations suggested that variant was likely to cause aberrant splicing (Table S3). To evaluate the pathogenicity of the variant, we performed the exon trapping assay and found that the variant caused skipping of exon 6 (Fig. 2b, c). The skipping of the 95-bp exon induces a frame-shift that generates a new stop codon in all five known transcript variants of TNFRSF11A (Fig. 3). For variants 1–3, the new stop codon resides before the penultimate exon, thus leading a nonsense-mediated mRNA decay (NMD). For transcript variants 4 and 5, the new stop codon resides less than 50 nucleotides upstream of the last exon–exon junction and at the last exon, respectively, where NMD would not happen according to the position rule [17]. Without NMD, the transcript variants 4 and 5 would be predicted to translate into the corresponding truncated proteins, p.N174Kfs*31 and p.N174Kfs*8 (Fig. 3).
Fig. 3
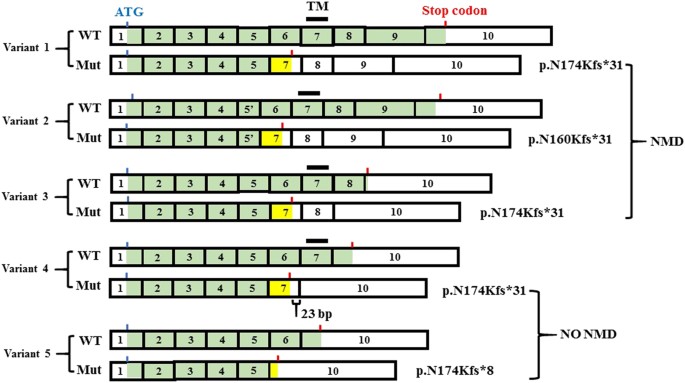
The alterative splicing of the TNFRSF11A gene and the effect of c.616+3A>G mutation on the isoforms. Five transcript variants are recorded in NCBI database (variant 1: NM_003839.3; variant 2: NM_001278268.1, variant 3: NM_001270949.1, variant 3: NM_001270950.1, variant 3: NM_001270951.1). The exons are numbered according to the longest variant (variant 1). Exon 5′ in variant 2 has a 42 bp in-frame deletion in comparison with exon 5. Coding regions are marked in green. The blue and red bars show the positions of the first ATG and the stop codons, respectively; the black bar shows the transmembrane domain (TM). WT wild type transcript, Mut mutant transcript. In every transcript variant, exon 6 skipping causes a frame-shift generating a new stop codon. For variants 1–3, the new stop codons reside before the penultimate exons, thus leading a nonsense-mediated mRNA decay (NMD). For variants 4 and 5, the new stop codons reside <50 nucleotides upstream of the last exon–exon junction and at the last exon, respectively, thus not causing NMD. TM was present in the isoform from the wild type variant 4, but absent in that from the mutant variant 4. Both isoforms lacked the intra-cellular signal transduction regions
Thus, by using a whole-exome sequencing, we identified TNFRSF11A as the 2nd disease gene for DOS. SLC29A3 has been reported as the disease gene for DOS [5]. Mutations of SLC29A3 also cause histiocytosis-lymphadenopathy plus syndrome (MIM 602782), a group of autosomal recessive conditions with little or no skeletal involvement [18, 19]. SLC29A3 −/− mice developed a spontaneous and progressive macrophage-dominated histiocytosis, which recapitulated the manifestations of histiocytosis-lymphadenopathy plus syndrome caused by SLC29A3 mutations [6]; however, the mice did not present overt skeletal dysplasia including osteosclerosis and platyspondyly, the main characteristics of DOS [6]. Thus, the role of SLC29A3 in DOS needs to be decided carefully in the further study.
TNFRSF11A (RANK) is the receptor for RANK-Ligand (RANKL) and part of the RANK/RANKL/OPG signaling pathway that regulates osteoclast differentiation and activation. RANKL expressed by osteoblasts and stromal stem cells binds to its receptor, RANK, on the surface of osteoclasts and their precursors, activate the downstream pathway. This pathway promotes the precursors differentiation into multinucleated osteoclasts, activates osteoclast and facilitates osteoclast survival, finally contributing to the increased bone resorption [20]. TNFRSF11A mutations have been reported in familial expansile osteolysis (MIM 174810), Paget disease of bone 2, early-onset (MIM 602080), and osteopetrosis, autosomal recessive 7 (OP-AR7; MIM 612301). The former two are of dominant inheritance, and the last one is of recessive inheritance. All the mutations identified in familial expansile osteolysis were in-frame insertions and led to increased constitutive RANK signaling [21]. In contrast, the mutations found in OP-AR7 are mostly definite loss of function mutations, which generate premature stop codon leading to nonsense mutation mediated mRNA decay (NMD) [22, 23]. The results suggest that the dominant RANK disorders characterized by enhanced bone resorption were caused by increased RANK function while OP-AR7 were caused by loss-of-function. DOS is clearly differentiated from OP-AR7 by the platyspondyly with punctate densities. Twelve TNFRSF11A mutations have been reported in OP-AR7 [22, 23]. The mutation, c.616+3A>G identified in our study is the 1st splice-site mutation in TNFRSF11A.
TNFRSF11A has five alternative splicing variants (Fig. 3), which produce five different protein isoforms. The significance of the alternative splicing and resultant isoforms remain unknown. They may have different expression patterns spatially and temporally and hence lead to different function. In all isoforms, the N-terminal receptor domain is almost intact, while the intra-cellular signal transduction region has many defects in the isoforms produced from variants 3–5 and the transmembrane domain is lost in that from variant 5 (Fig. 3). Previously, five stop-codon producing mutations (nonsense or frame-shift mutations) have been reported in TNFRSF11A in the patients with OP-AR7 (Table 1). All these mutations are not considered to affect on the isoform structure; they are predicted to result in the wild-type isoforms or produce no isoform due to NMD. In contrast, the splicing mutation we identified in this study could cause different effects on the alternative transcripts (Table 1). While the mutant splicing variants 1–3 would be considered subjected to NMD, the mutant variant 4 and 5 would escape from NMD, producing the truncated mutant RANK proteins (Fig. 3). Interestingly, while the isoform produced from the wild-type variant 4 has the transmembrane domain, that from the mutant variant 4 lacks the transmembrane domain and its extracellular domain is altered. The complexity of the mutation effects on the splicing probably contributes to the unique phenotypes of DOS. Identification of additional TNFRSF11A mutations and the close evaluation of the genotype–phenotype relation of DOS and OP-AR7 would give us the insight into the significance of the alternative splicing and the role of TNFRSF11A in the development and maintenance of the skeleton.
Table 1 Effects of the stop-codon producing mutations of TNFRSF11A on five isoforms
References
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167:2869–92.
Article CAS Google Scholar - Houston CS, Gerrard JW, Ives EJ. Dysosteosclerosis. Am J Roentgenol. 1978;130:988–91.
Article CAS Google Scholar - Whyte MP, Wenkert D, McAlister WH, Novack DV, Nenninger AR, Zhang X, et al. Dysosteosclerosis presents as an ‘Osteoclast-Poor’ form of osteopetrosis: Comprehensive investigation of a 3-year-old girl and literature review. J Bone Miner Res. 2010;25:2527–39.
Article Google Scholar - Elçioglu NH, Vellodi A, Hall CM. Dysosteosclerosis: a report of three new cases and evolution of the radiological findings. J Med Genet. 2002;39:603–7.
Article Google Scholar - Campeau PM, Lu JT, Sule G, Jiang MM, Bae Y, Madan S, et al. Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum Mol Genet. 2012;21:4904–9.
Article CAS Google Scholar - Hsu CL, Lin W, Seshasayee D, Chen YH, Ding X, Lin Z, et al. Equilibrative nucleoside transporter 3 deficiency perturbs lysosome function and macrophage homeostasis. Science. 2012;335:89–92.
Article CAS Google Scholar - Guo L, Elcioglu NH, Wang Z, Demirkol YK, Isguven P, Matsumoto N, et al. Novel and recurrent COL11A1 and COL2A1 mutations in the Marshall–Stickler syndrome spectrum. Hum Genome Var. 2017;4:17040.
Article CAS Google Scholar - Guo L, Elcioglu NH, Mizumoto S, Wang Z, Noyan B, Albayrak HM, et al. Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. J Hum Genet. 2017;62:797–801.
Article CAS Google Scholar - Guo L, Elcioglu NH, Iida A, Demirkol YK, Aras S, Matsumoto N, et al. Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J Hum Genet. 2017;62:447–51.
Article CAS Google Scholar - Guo L, Girisha KM, Iida A, Hebbar M, Shukla A, Shah H, et al. Identification of a novel LRRK1 mutation in a family with osteosclerotic metaphyseal dysplasia. J Hum Genet. 2017;62:437–41.
Article CAS Google Scholar - Wang Z, Horemuzova E, Iida A, Guo L, Liu Y, Matsumoto N, et al. Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J Hum Genet. 2017;62:503–6.
Article CAS Google Scholar - McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Article CAS Google Scholar - Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Article Google Scholar - Miyatake S, Tada H, Moriya S, Takanashi J, Hirano Y, Hayashi M, et al. Atypical giant axonal neuropathy arising from a homozygous mutation by uniparental isodisomy. Clin Genet. 2015;87:395–7.
Article CAS Google Scholar - Furuichi T, Kayserili H, Hiraoka S, Nishimura G, Ohashi H, Alanay Y, et al. Identification of loss-of-function mutations of SLC35D1 in patients with Schneckenbecken dysplasia, but not with other severe spondylodysplastic dysplasias group diseases. J Med Genet. 2009;46:562–8.
Article CAS Google Scholar - Nakashima Y, Sakamoto Y, Nishimura G, Ikegawa S, Iwamoto Y. A novel type II collagen gene mutation in a family with spondyloepiphyseal dysplasia and extensive intrafamilial phenotypic diversity. Hum Genome Var. 2016;3:16007.
Article CAS Google Scholar - Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. 1998;23:198–9.
Article CAS Google Scholar - Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu LA, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. 2008;83:529–34.
Article CAS Google Scholar - Spiegel R, Cliffe ST, Buckley MF, Crow YJ, Urquhart J, Horovitz Y, et al. Expanding the clinical spectrum of SLC29A3 gene defects. Eur J Med Genet. 2010;53:309–13.
Article Google Scholar - Boyce BF, Xing L. The RANKL/RANK/OPG pathway. Curr Osteoporos Rep. 2007;5:98–104.
Article Google Scholar - Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–48.
Article CAS Google Scholar - Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76.
Article CAS Google Scholar - Pangrazio A, Cassani B, Guerrini MM, Crockett JC, Marrella V, Zammataro L, et al. RANK-dependent autosomal recessive osteopetrosis: characterization of five new cases with novel mutations. J Bone Miner Res. 2012;27:342–51.
Article CAS Google Scholar
Acknowledgements
We would like to thank the patient and her family for their help to the study.
Funding
This study was supported by research grants from Japan Agency For Medical Research and Development (AMED) (Contract No. 14525125), the Japan Society for the Promotion of Science (WAKATE B, No. 17K16710), and RIKEN-MOST.
Author information
Author notes
- These authors contributed equally: Long Guo, Nursel H. Elcioglu.
Authors and Affiliations
- Laboratory for Bone and Joint Diseases, RIKEN Center for Integrative Medical Sciences, Tokyo, 108-8639, Japan
Long Guo, Zheng Wang & Shiro Ikegawa - Department of Pediatric Genetics, Marmara University Medical School, 34890, Istanbul, Turkey
Nursel H. Elcioglu & Ozge K. Karalar - Eastern Mediterranean University Medical School, Mersin 10, Cyprus, Turkey
Nursel H. Elcioglu - Department of Orthopedics, Marmara University Medical School, 34890, Istanbul, Turkey
Mert O. Topkar - Department of Medical Genetics, Institute of Basic Medical Sciences, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
Zheng Wang - Department of Human Genetics, Yokohama City University Graduate School of Medicine, Yokohama, 236-0004, Japan
Naomichi Matsumoto & Noriko Miyake - Department of Pediatric Imaging, Tokyo Metropolitan Children’s Medical Center, Fuchu, 183-8561, Japan
Gen Nishimura
Authors
- Long Guo
You can also search for this author inPubMed Google Scholar - Nursel H. Elcioglu
You can also search for this author inPubMed Google Scholar - Ozge K. Karalar
You can also search for this author inPubMed Google Scholar - Mert O. Topkar
You can also search for this author inPubMed Google Scholar - Zheng Wang
You can also search for this author inPubMed Google Scholar - Yuma Sakamoto
You can also search for this author inPubMed Google Scholar - Naomichi Matsumoto
You can also search for this author inPubMed Google Scholar - Noriko Miyake
You can also search for this author inPubMed Google Scholar - Gen Nishimura
You can also search for this author inPubMed Google Scholar - Shiro Ikegawa
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toShiro Ikegawa.
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Guo, L., Elcioglu, N.H., Karalar, O.K. et al. Dysosteosclerosis is also caused by TNFRSF11A mutation.J Hum Genet 63, 769–774 (2018). https://doi.org/10.1038/s10038-018-0447-6
- Received: 15 February 2018
- Revised: 06 March 2018
- Accepted: 07 March 2018
- Published: 22 March 2018
- Issue Date: June 2018
- DOI: https://doi.org/10.1038/s10038-018-0447-6