The evolutionary history of vertebrate RNA viruses (original) (raw)
- Article
- Published: 04 April 2018
- Xian-Dan Lin4 na1,
- Xiao Chen5 na1,
- Jun-Hua Tian6 na1,
- Liang-Jun Chen1,
- Kun Li1,
- Wen Wang1,
- John-Sebastian Eden3,
- Jin-Jin Shen7,
- Li Liu5,
- Edward C. Holmes1,2,3 &
- …
- Yong-Zhen Zhang1,2
Nature volume 556, pages 197–202 (2018)Cite this article
- 45k Accesses
- 785 Citations
- 544 Altmetric
- Metrics details
Subjects
This article has been updated
Abstract
Our understanding of the diversity and evolution of vertebrate RNA viruses is largely limited to those found in mammalian and avian hosts and associated with overt disease. Here, using a large-scale meta-transcriptomic approach, we discover 214 vertebrate-associated viruses in reptiles, amphibians, lungfish, ray-finned fish, cartilaginous fish and jawless fish. The newly discovered viruses appear in every family or genus of RNA virus associated with vertebrate infection, including those containing human pathogens such as influenza virus, the Arenaviridae and Filoviridae families, and have branching orders that broadly reflected the phylogenetic history of their hosts. We establish a long evolutionary history for most groups of vertebrate RNA virus, and support this by evaluating evolutionary timescales using dated orthologous endogenous virus elements. We also identify new vertebrate-specific RNA viruses and genome architectures, and re-evaluate the evolution of vector-borne RNA viruses. In summary, this study reveals diverse virus–host associations across the entire evolutionary history of the vertebrates.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout
Additional access options:
Fig. 1: Identification of vertebrate-associated viruses in divergent vertebrate host groups.
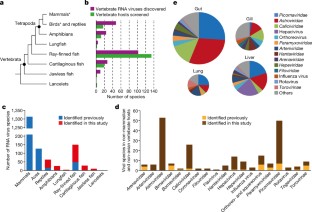
The alternative text for this image may have been generated using AI.
Fig. 2: Evolutionary history of 17 major vertebrate-specific virus families or genera.
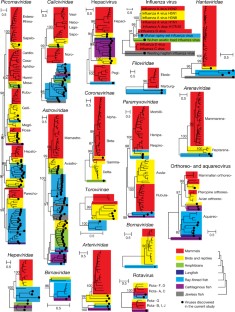
The alternative text for this image may have been generated using AI.
Fig. 3: Long-term evolutionary relationships between vertebrate hosts and their associated viruses.
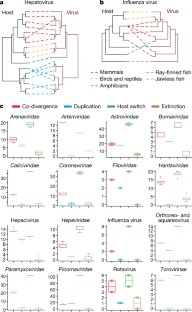
The alternative text for this image may have been generated using AI.
Fig. 4: Evaluating the timescale of vertebrate virus evolution using EVEs.

The alternative text for this image may have been generated using AI.
Fig. 5: Evolution of vertebrate-associated virus genomes.

The alternative text for this image may have been generated using AI.
Similar content being viewed by others
Change history
26 June 2018
Change history: In this Article, author Li Liu should be associated with affiliation number 5 (College of Marine Sciences, South China Agricultural University, Guangzhou, Guangdong, China), rather than affiliation number 4 (Wenzhou Center for Disease Control and Prevention, Wenzhou, Zhejiang, China). This has been corrected online.
References
- Shi, M. et al. Redefining the invertebrate RNA virosphere. Nature 540, 539–543 (2016).
Article ADS CAS Google Scholar - King, A. M. Q., Adams, M. J., Carstens, E. B. & Lefkowitz, E. J. Virus Taxonomy: 9th Report of the International Committee on Taxonomy of Viruses (Elsevier Academic, Amsterdam, 2012).
- Essbauer, S. & Ahne, W. Viruses of lower vertebrates. J. Vet. Med. B Infect. Dis. Vet. Public Health 48, 403–475 (2001).
Article PubMed CAS Google Scholar - Batts, W., Yun, S., Hedrick, R. & Winton, J. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res. 158, 116–123 (2011).
Article PubMed CAS Google Scholar - Mikalsen, A. B. et al. Characterization of a novel calicivirus causing systemic infection in atlantic salmon (Salmo salar L.): proposal for a new genus of caliciviridae. PLoS ONE 9, e107132 (2014).
Article ADS PubMed PubMed Central CAS Google Scholar - Shi, M. et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 90, 659–669 (2015).
Article PubMed PubMed Central CAS Google Scholar - Stenglein, M. D. et al. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. MBio 3, e00180–12 (2012).
Article PubMed PubMed Central CAS Google Scholar - Hedges, S. B., Marin, J., Suleski, M., Paymer, M. & Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol 32, 835–845 (2015).
Article PubMed PubMed Central CAS Google Scholar - Dill, J. A. et al. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J. Virol. 90, 7920–7933 (2016).
Article PubMed PubMed Central CAS Google Scholar - Wang, T. H., Donaldson, Y. K., Brettle, R. P., Bell, J. E. & Simmonds, P. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 75, 11686–11699 (2001).
Article PubMed PubMed Central CAS Google Scholar - Brinkmann, H., Venkatesh, B., Brenner, S. & Meyer, A. Nuclear protein-coding genes support lungfish and not the coelacanth as the closest living relatives of land vertebrates. Proc. Natl Acad. Sci. USA 101, 4900–4905 (2004).
Article ADS PubMed PubMed Central CAS Google Scholar - Conow, C., Fielder, D., Ovadia, Y. & Libeskind-Hadas, R. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 5, 16 (2010).
Article PubMed PubMed Central Google Scholar - Geoghegan, J. L., Duchêne, S. & Holmes, E. C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 13, e1006215 (2017).
Article PubMed PubMed Central CAS Google Scholar - Charleston, M. A. & Robertson, D. L. Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst. Biol 51, 528–535 (2002).
Article PubMed CAS Google Scholar - de Vienne, D. M. et al. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol. 198, 347–385 (2013).
Article PubMed Google Scholar - Wertheim, J. O. & Kosakovsky Pond, S. L. Purifying selection can obscure the ancient age of viral lineages. Mol. Biol. Evol 28, 3355–3365 (2011).
Article PubMed PubMed Central CAS Google Scholar - Zhang, Y. Z. & Holmes, E. C. What is the time-scale of hantavirus evolution? Infect. Genet. Evol. 25, 144–145 (2014).
Article PubMed Google Scholar - Katzourakis, A. & Gifford, R. J. Endogenous viral elements in animal genomes. PLoS Genet. 6, e1001191 (2010).
Article PubMed PubMed Central CAS Google Scholar - Taylor, D. J., Leach, R. W. & Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 10, 193 (2010).
Article PubMed PubMed Central CAS Google Scholar - Horie, M. et al. Endogenous non-retroviral RNA virus elements in mammalian genomes. Nature 463, 84–87 (2010).
Article ADS PubMed PubMed Central CAS Google Scholar - Li, C. X. et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 4, e05378 (2015).
- Longdon, B. et al. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 1, vev014 (2015).
Article PubMed PubMed Central Google Scholar - Qin, X. C. et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl Acad. Sci. USA 111, 6744–6749 (2014).
Article ADS PubMed PubMed Central CAS Google Scholar - Kelly, A. G., Netzler, N. E. & White, P. A. Ancient recombination events and the origins of hepatitis E virus. BMC Evol. Biol. 16, 210 (2016).
Article PubMed PubMed Central CAS Google Scholar - Han, G. Z. & Worobey, M. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog. 8, e1002790 (2012).
Article PubMed PubMed Central CAS Google Scholar - Aiewsakun, P. & Katzourakis, A. Marine origin of retroviruses in the early Palaeozoic Era. Nat. Commun. 8, 13954 (2017).
Article ADS PubMed PubMed Central CAS Google Scholar - Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Article PubMed PubMed Central CAS Google Scholar - Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Article PubMed PubMed Central CAS Google Scholar - Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Article PubMed PubMed Central CAS Google Scholar - Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Article PubMed PubMed Central Google Scholar - Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Article PubMed PubMed Central CAS Google Scholar - Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Article PubMed PubMed Central CAS Google Scholar - Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Article PubMed PubMed Central CAS Google Scholar - Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol 52, 696–704 (2003).
Article PubMed Google Scholar - Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Article PubMed CAS Google Scholar - Parker, J., Rambaut, A. & Pybus, O. G. Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246 (2008).
Article PubMed CAS Google Scholar - Betancur-R, R. et al. The tree of life and a new classification of bony fishes. PLoS Curr. 5, https://doi.org/10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288 (2013).
Acknowledgements
This study was supported by the Special National Project on Research and Development of Key Biosafety Technologies (2016YFC1201900, 2016YFC1200101) and the National Natural Science Foundation of China (Grants 81672057, 81611130073). E.C.H. and M.S. are funded by an ARC Australian Laureate Fellowship to E.C.H. (FL170100022). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank students at the Zoonosis branch of the China CDC, especially W.-C. Wu, J.-W. Shao, C.-X. Li, J.-J. Guo and K.-L. Song for assistance with virus and host sequence confirmation, and we thank B. Yu for help with the collection of animal samples. We acknowledge the University of Sydney high-performance computing (HPC) service at The University of Sydney for providing resources that have contributed to the research results reported within this paper
Reviewer information
Nature thanks A. Rambaut and M. Worobey for their contribution to the peer review of this work.
Author information
Author notes
- These authors contributed equally: Mang Shi, Xian-Dan Lin, Xiao Chen, Jun-Hua Tian.
Authors and Affiliations
- State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Mang Shi, Liang-Jun Chen, Kun Li, Wen Wang, Edward C. Holmes & Yong-Zhen Zhang - Shanghai Public Health Clinical Center & Institute of Biomedical Sciences, Fudan University, Shanghai, China
Mang Shi, Edward C. Holmes & Yong-Zhen Zhang - Marie Bashir Institute for Infectious Diseases and Biosecurity, Charles Perkins Centre, School of Life and Environmental Sciences and Sydney Medical School, The University of Sydney, Sydney, New South Wales, Australia
Mang Shi, John-Sebastian Eden & Edward C. Holmes - Wenzhou Center for Disease Control and Prevention, Wenzhou, China
Xian-Dan Lin - College of Marine Sciences, South China Agricultural University, Guangzhou, China
Xiao Chen & Li Liu - Wuhan Center for Disease Control and Prevention, Wuhan, China
Jun-Hua Tian - Yancheng Center for Disease Control and Prevention, Yancheng, China
Jin-Jin Shen
Authors
- Mang Shi
- Xian-Dan Lin
- Xiao Chen
- Jun-Hua Tian
- Liang-Jun Chen
- Kun Li
- Wen Wang
- John-Sebastian Eden
- Jin-Jin Shen
- Li Liu
- Edward C. Holmes
- Yong-Zhen Zhang
Contributions
M.S. and Y.-Z.Z. conceived and designed the study. M.S., X.-D.L., X.C., J.-H.T., K.L., L.-J.C., J.-J.S., L.L. and Y.-Z.Z. organized field work, and collected samples. M.S., X.-D.L., X.C., J.-H.T., K.L., L.-J.C., W.W., J.-J.S., L.L. and Y.-Z.Z. performed experiments. M.S., J.-S.E., E.C.H. and Y.-Z.Z. analysed data. M.S., E.C.H. and Y.-Z.Z. wrote the paper with input from all authors. Y.-Z.Z. led the study.
Corresponding author
Correspondence toYong-Zhen Zhang.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Phylogenetic positions of vertebrate-associated positive-sense and double-stranded RNA viruses within the broader diversity of RNA viruses.
Phylogenies were estimated using a maximum likelihood method and midpoint-rooted for clarity only. Viruses discovered here are labelled with solid black circles. The name of the major clade (phylogeny) is shown at the top of each tree, and taxonomic names are shown to the right. The vertebrate associated virus diversity is shaded in grey. All horizontal branch lengths are scaled to the number of amino acid substitutions per site.
Extended Data Fig. 2 Phylogenetic positions of vertebrate-associated negative-sense RNA viruses within the broader diversity of RNA viruses.
Phylogenies were estimated using a maximum likelihood method and midpoint-rooted for clarity only. Viruses discovered here are labelled with solid black circles. The name of the major clade (phylogeny) is shown at the top of each tree, and taxonomic names are shown to the right. The vertebrate associated virus diversity is shaded in grey. All horizontal branch lengths are scaled to the number of amino acid substitutions per site.
Extended Data Fig. 3 The phylogenies of potentially new families of vertebrate-associated viruses.
Viruses identified from vertebrate hosts are shaded with different colours. Sequences recovered from the Transcriptome Shotgun Assembly (TSA) database are marked with solid black diamonds, while those recovered from the Whole-Genome Shotgun (WGS) contigs database (that is, endogenous virus elements) are marked with open triangles. For vertebrate viruses, the relevant taxonomic and tissue information is provided in the sequence names.
Extended Data Fig. 4 Evolutionary history of four groups of vector-borne RNA virus.
Each phylogenetic tree was estimated using a maximum likelihood method. Within each phylogeny, the viruses newly identified here are marked with solid black circles, the vertebrate host groups are indicated by different colours, and the vector symbol is shown next to viruses known to be transmitted by vectors. The name of the virus family or genus is shown at the top of each phylogeny, and the lower level virus taxonomic names are shown to the right.
Extended Data Fig. 5 Evolution of vertebrate-associated negative-sense RNA virus genomes.
Representative genomes from negative-sense RNA virus families/genera are shown. The regions that encode major functional proteins or protein domains are labelled on each of the genomes. Homologous regions within each family are connected with orange dotted lines. Schematic phylogenetic relationships are shown next to the genomes diagrams. Coverage plots are shown underneath novel genome structures. Reverse-complementary sequences are shown for negative-sense RNA viruses with complete termini. A Sanger sequencing chromatogram is shown at a GC-rich hairpin-forming region of the Wenling frogfish arenavirus 2 genome, in which the coverage drops substantially. Host associations are labelled to the right of tree using solid circles with different colours. Host associations and abbreviation of functional domains are described at the bottom of the figure.
Extended Data Fig. 6 Evolution of vertebrate-associated positive-sense RNA virus genomes.
Representative genomes from positive-sense RNA virus families or genera are shown. The regions that encode major functional proteins or protein domains are labelled on each of the genomes. Homologous regions within or between viral families are connected by orange dotted lines. Host associations are reflected in the colour of the virus names. Host association colour schemes and the abbreviations of functional domains are described at the bottom of the figure.
Supplementary information
Rights and permissions
About this article
Cite this article
Shi, M., Lin, XD., Chen, X. et al. The evolutionary history of vertebrate RNA viruses.Nature 556, 197–202 (2018). https://doi.org/10.1038/s41586-018-0012-7
- Received: 15 September 2017
- Accepted: 23 February 2018
- Published: 04 April 2018
- Version of record: 04 April 2018
- Issue date: 12 April 2018
- DOI: https://doi.org/10.1038/s41586-018-0012-7