Sex differences in immune responses that underlie COVID-19 disease outcomes (original) (raw)
Main
SARS-CoV-2 is the novel coronavirus first detected in Wuhan, China, in November 2019 that causes COVID-196. On 11 March 2020, the World Health Organization (WHO) declared COVID-19 a pandemic[7](/articles/s41586-020-2700-3#ref-CR7 "WHO. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19 - 11 March 2020 https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020
(2020)."). A growing body of evidence reveals that male sex is a risk factor for a more severe disease, including death. Globally, approximately 60% of deaths from COVID-19 are reported in men[5](/articles/s41586-020-2700-3#ref-CR5 "Gebhard, C., Regitz-Zagrosek, V., Neuhauser, H. K., Morgan, R. & Klein, S. L. Impact of sex and gender on COVID-19 outcomes in Europe. Biol. Sex Differ. 11, 29 (2020)."), and a cohort study of 17 million adults in England reported a strong association between male sex and the risk of death from COVID-19 (hazard ratio 1.59, 95% confidence interval 1.53–1.65)[8](/articles/s41586-020-2700-3#ref-CR8 "Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020).").Past studies have shown that sex has a considerable effect on the outcome of infections and has been associated with underlying differences in immune responses to infection9,10. For example, the prevalence of hepatitis A and tuberculosis are notably higher in men that in women11. Viral loads are consistently higher in male patients with hepatitis C virus and human immunodeficiency virus (HIV)12,13. By contrast, women mount a more robust immune response to vaccines14. These findings collectively suggest a more robust ability among women to control infectious agents. However, the mechanism by which SARS-CoV-2 causes more severe disease in male patients than in female patients remains unknown.
To determine the immune responses against SARS-CoV-2 infection in male and female patients, we performed detailed analyses on the sex differences in immune phenotypes by the assessment of viral loads, levels of SARS-CoV-2-specific antibodies, plasma cytokines or chemokines, and blood-cell phenotypes.
Overview of the study design
Patients who were admitted to the Yale-New Haven Hospital between 18 March and 9 May 2020 and were confirmed positive for SARS-CoV-2 by RT–PCR from nasopharyngeal and/or oropharyngeal swabs in CLIA-certified laboratories were enrolled through the IMPACT biorepository study15. In the IMPACT study, biospecimens including blood, nasopharyngeal swabs, saliva, urine and stool samples were collected at study enrolment (baseline denotes the first time point) and longitudinally on average every 3 to 7 days (serial time points). The detailed demographics and clinical characteristics of these 98 participants are shown in Extended Data Table 1. Plasma and peripheral blood mononuclear cells (PBMCs) were isolated from whole blood, and plasma was used for titre measurements of SARS-CoV-2 spike S1 protein-specific IgG and IgM antibodies (anti-S1-IgG and -IgM) and cytokine or chemokine measurements. Freshly isolated PBMCs were stained and analysed by flow cytometry15. We obtained longitudinal serial time-point samples from a subset of these 98 study participants (n = 48; information in Extended Data Table 1). To compare the immune phenotypes between sexes, two sets of data analyses were performed in parallel—baseline and longitudinal, as described below. As a control group, healthcare workers (HCWs) from Yale-New Haven Hospital were enrolled who were uninfected with COVID-19. Demographics and background information for the HCW group and the demographics of HCWs for cytokine assays and flow cytometry assays for the primary analyses are in Extended Data Table 1. Demographic data, time-point information of the samples defined by the days from the symptom onset (DFSO) in each patient, treatment information, and raw data used to generate figures and tables is in Supplementary Table 1.
Baseline analysis
The baseline analysis was performed on samples from the first time point of patients who met the following criteria: not in intensive care unit (ICU), had not received tocilizumab, and had not received high doses of corticosteroids (prednisone equivalent of more than 40 mg) before the first sample collection date. This patient group, cohort A, consisted of 39 patients (17 male and 22 female) (Extended Data Tables 1, 2). Intersex and transgender individuals were not represented in this study. Figures 1–4 represent analyses of baseline raw values obtained from patients in cohort A. In cohort A patients, male and female patients were matched in terms of age, body mass index (BMI), and DFSO at the first time point sample collection (Extended Data Fig. 1a). However, there were significant differences in age and BMI between HCW controls and patients (patients had higher age and BMI values) (Extended Data Table 1), and therefore an age- and BMI-adjusted difference-in-differences analysis was also performed in parallel (Extended Data Table 3).
Fig. 1: Comparison of viral RNA concentrations, titres of anti-SARS-CoV-2 antibodies, and plasma cytokines and chemokine levels at the first sampling of cohort A patients.
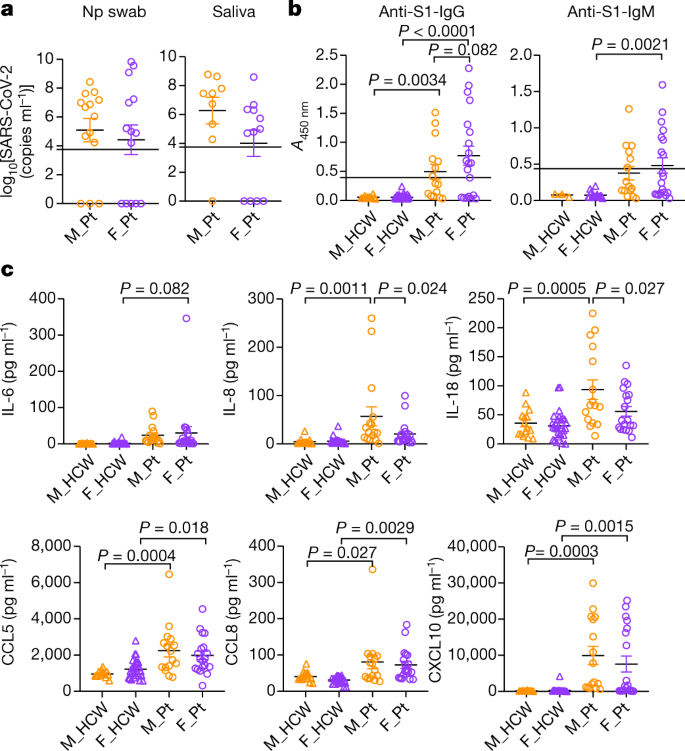
a, Comparison of viral RNA measured from nasopharyngeal (Np) swab and saliva. n = 14 for male and female patients (M_Pt and F_Pt, respectively) for nasopharyngeal samples, and n = 9 and 12, respectively, for saliva samples. Dotted lines indicate the detection limit of the assay (5,610 copies ml−1), and negatively tested data are shown on the x axis. ND, not detected. b, Titres of specific IgG and IgM antibodies against SARS-CoV-2 S1 protein were measured. n = 13, 74, 15 and 20 for IgG, and n = 3, 18, 15 and 20 for IgM, for male HCW (M_HCW), female HCW (F_HCW), M_Pt and F_Pt, respectively. The cut-off values for positivity are shown by the dotted lines. c, Comparison of the plasma levels of representative innate immune cytokines and chemokines. n = 15, 28, 16 and 19 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. P values were determined by unpaired two-tailed _t_-test (a) or one-way analysis of variance (ANOVA) with Bonferroni multiple comparison test (b, c). All P values < 0.10 are shown. Data are mean ± s.e.m. The results of all the cytokines or chemokines measured can be found in Extended Data Fig. 1b.
Fig. 2: Differences in composition of PBMCs between male and female patients in cohort A at the first sampling.
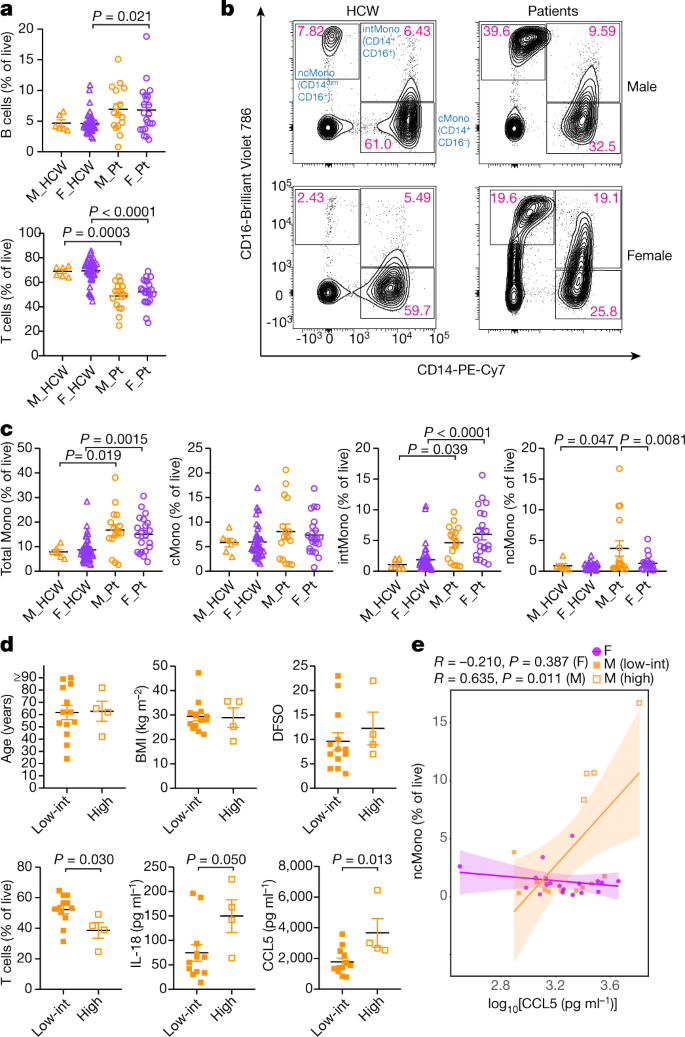
a, Comparison on the proportion of B cells (top) and T cells (bottom) in live PBMCs. n = 6, 42, 16 and 21 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. b, Representative 2D plots for CD14 and CD16 in monocytes gate (live/singlets/CD19−CD3−/CD56−CD66b−). Numbers in red indicate the percentages of each population in the parent monocyte gate. c, Comparison between percentages of total monocytes, classical monocytes (cMono), intermediate monocytes (intMono) and non-classical monocytes (ncMono) in the live PBMCs. n = 6, 42, 16 and 21 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. d, Comparison of age, BMI, DFSO, T cells (percentage of live PBMCs) and plasma IL-18 and CCL5 levels between male patients who had high non-classical monocytes and low-intermediate non-classical monocytes. n = 13 and 4 for ‘low-int’ and ‘high’ group, respectively, for age, BMI and DFSO. n = 12 and 4 for ‘low-int’ and ‘high’ group, respectively, for T cells and IL-18 or CCL5 levels. e, Correlation between plasma CCL5 levels and non-classical monocytes (percentage of live cells). Pearson correlation coefficients (R) and P values for each sex are shown. Lines represent linear regression lines and shading represents 95% confidence intervals for each sex. ncMono-high male patients (n = 4) are shown with orange open squares, and ncMono-low-int male patients (n = 11) are shown with orange closed squares. n = 19 for female patients (purple circles). Data are mean ± s.e.m. in a, c and d. P values were determined by one-way ANOVA with Bonferroni multiple comparison test (a, c) or unpaired two-tailed _t_-test (d). All P values < 0.10 are shown.
Fig. 3: Sex difference in T cell phenotype at the first sampling of cohort A patients.
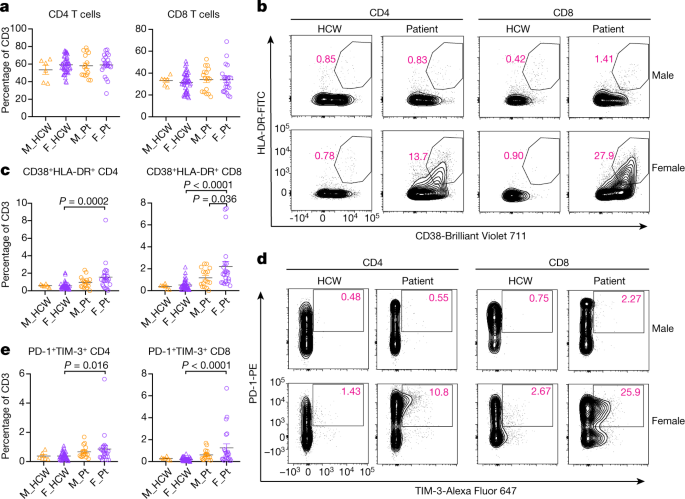
a, Percentages of CD4 and CD8 in the CD3-positive cells. b, Representative 2D plots for CD38 and HLA-DR in the CD4 and CD8 T cells. Numbers in red indicate the percentages of CD38+HLA-DR+ populations in the parent gate (live/singlets/CD3+/CD4+ or CD8+). c, Percentages of CD38+HLA-DR+ CD4 or CD8 cells in CD3-positive cells. d, Representative 2D plots for PD-1 and TIM-3 in the CD4 and CD8 T cells. Numbers in red indicate the percentages of PD-1+TIM-3+ populations in the parent gate (live/singlets/CD3+/CD4+ or CD8+/CD45RA−). e, Percentages of PD-1+TIM-3+ CD4 or CD8 cells in CD3-positive cells. n = 6, 45, 16 and 22 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. P values were determined by one-way ANOVA with Bonferroni multiple comparison test. Data are mean ± s.e.m. All P values < 0.10 are shown.
Fig. 4: Differential immune phenotypes at the first sampling and disease progression between sexes in cohort A patients.
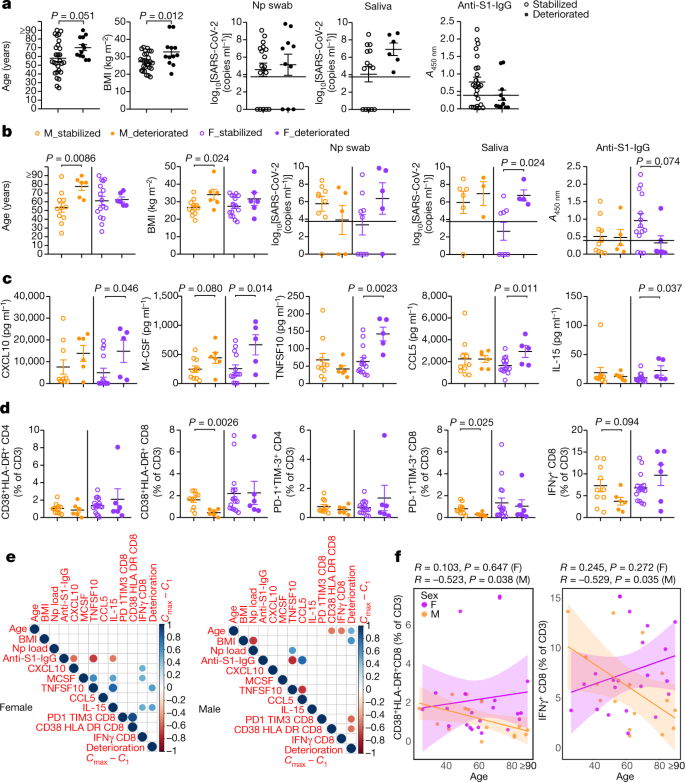
a, b, Sex-aggregated (a) and sex-disaggregated (b) comparison of age, BMI, RNA concentration in nasopharyngeal swab and saliva, and anti-S1-IgG antibodies between the stabilized and deteriorated group. n = 11, 6, 16 and 6 for age and BMI, n = 9, 5, 9 and 5 for nasopharyngeal swab, n = 6, 3, 8 and 4 for saliva, and n = 10, 5, 14 and 6 for anti-S1-IgG antibodies, for M_stabilized, M_deteriorated, F_stabilized and F_deteriorated group, respectively. Dotted lines in the viral concentration and anti-S1-IgG panels indicate the detection limit and cut-off value for positivity, respectively. c, Cytokine or chemokine comparison between stabilized and deteriorated groups. n = 10, 6, 14 and 5 for the M_stabilized, M_deteriorated, F_stabilized and F_deteriorated groups, respectively. d, Comparisons in the proportions of activated (CD38+HLA-DR+) and terminally differentiated (PD-1+TIM-3+) CD4 or CD8 T cells, and IFNγ+CD8 T cells in CD3-positive T cells are shown. n = 10, 6, 16 and 6 for M_stabilized, M_deteriorated, F_stabilized and F_deteriorated group, respectively. e, Pearson correlation heat maps of the indicated parameters are shown for each sex. For viral RNA concentrations and cytokine or chemokine levels, log-transformed values were used for the calculation of the correlations. The size and colour of the circles indicate the correlation coefficient (R), and only statistically significant correlations (P < 0.05) are shown. Clinical deterioration from the first time point was scored by _C_max − _C_1. n = 17 and 22 for male and female, respectively. f, Correlation between age and CD38+HLA-DR+ CD8 T cells (left) and IFNγ+CD8 T cells (right). Pearson correlation coefficient (R) and P values for each correlation and sex are shown. Lines represent linear regression lines and shading represents 95% confidence intervals for each sex. P values were determined by unpaired two-tailed _t_-test in a–d. Data are mean ± s.e.m. All P values < 0.10 are shown.
Longitudinal analysis
As parallel secondary analyses, we performed longitudinal analysis on a total patient cohort (cohort B) to evaluate the difference in immune response over the course of the disease between male and female patients. Cohort B included all patient samples from cohort A (including several time-point samples from the cohort A patients) as well as an additional 59 patients who did not meet the inclusion criteria for cohort A. Because cohort B included more severely affected patients in ICU, the average clinical scores were higher in cohort B than in cohort A (mean ± s.d.: 1.3 ± 0.5 (female) and 1.4 ± 0.5 (male) for cohort A, and 2.5 ± 1.5 (female) and 2.7 ± 1.3 (male) for cohort B) (Extended Data Table 1). This analysis included several time-point samples from 98 participants in total. Data from cohort B were analysed for sex differences in immune responses among patients using longitudinal analysis, controlling for potential confounding by age, BMI, receipt of immunomodulatory treatment (tocilizumab or corticosteroids), DFSO and ICU status. Second, we conducted a longitudinal analysis that compared male and female patients with COVID-19 to male and female HCWs, controlling for age and BMI. Adjusted least square means difference over time in immune responses between male and female patients with COVID-19 (Extended Data Table 4) and adjusted least square means difference over time in immune responses between male and female patients with COVID-19 and male and female HCWs (Extended Data Table 5) were calculated.
Sex differences in cytokines and chemokines
We first compared the concentrations of viral RNA of male and female patients. For both cohorts A and B, there was no difference by sex in terms of the viral RNA concentrations in nasopharyngeal swab and saliva (Fig. 1a, Extended Data Tables 3, 4).
Anti-SARS-CoV-2 S1-specific IgG and IgM (anti-S1-IgG and -IgM) antibodies were comparable in infected male and female in cohort A (Fig. 1b) and in cohort B (Extended Data Tables 4, 5). Thus, at baseline and during the course of the disease, there were no clear differences in the amount of IgG or IgM generated against the S1 protein between male and female patients.
Next, we analysed the levels of 71 cytokines and chemokines in the plasma. Levels of many pro-inflammatory cytokines, chemokines and growth factors, including IL-1β, IL-6, IL-8, TNF, CCL2, CXCL10 and G-CSF, are increased in the plasma of patients with COVID-1916. In line with previous reports, levels of inflammatory cytokine or chemokine were generally higher in patients than in controls (Fig. 1c, Extended Data Figs. 1b, 2a, Extended Data Table 3). The levels of type-I, -II or -III interferon (IFN) were comparable between the sexes in cohort A (Extended Data Fig. 1b, Extended Data Table 3). However, we found higher levels of IFNα2 in female patients than in male patients in cohort B (Extended Data Table 4). The levels of many cytokines, chemokines and growth factors were increased in patients compared to HCWs in both sexes, and the levels between sexes were comparable (Fig. 1c, Extended Data Fig. 1b, Extended Data Table 3). However, levels of IL-8 and IL-18 were significantly higher in male patients than in female patients in cohort A (Fig. 1c). In age- and BMI-adjusted analyses of cohort A, we found that although IL-8 and IL-18 were no longer significantly higher among male patients than in to female patients, IL-8 and CXCL10 were significantly increased in male patients compared to male HCWs than in female patients compared to female HCWs (difference-in-differences, Extended Data Table 3). In adjusted analyses of cohort B, although we did not see significant sex differences in the levels of IL-8 and IL-18, we found significantly higher levels of CCL5 in male patients than in female patients over the course of the disease (Extended Data Table 4) and significantly increased levels of CCL5 in male patients compared to male HCWs than in female patients compared to female HCWs (Extended Data Table 5, difference-in-differences). These data indicated that, although levels of most of the innate inflammatory cytokines and chemokines were comparable, there were a few factors that are more robustly increased at the baseline (IL-8 and IL-18) and during the course of the disease (CCL5) in male patients than in female patients.
Monocyte differences by sex
Next, we examined the immune cell phenotype by flow cytometry. Freshly isolated PBMCs were stained with specific antibodies to identify T cells, B cells, natural killer T cells, natural killer cells, monocytes, macrophages and dendritic cells to investigate the composition of PBMCs (Extended Data Fig. 2b). Consistent with a previous report on a decrease in T cells in patients16, in cohort A, the proportion of T cells in the live cells was significantly lower in patients, whereas the proportion of B cells was higher in both male and female patients than in HCWs (Fig. 2a, Extended Data Table 3). There was no difference in the numbers of B cells across all groups, but the numbers of T cells were lower in patients of both sexes (data not shown). By contrast, in cohort B, we found that male patients had significantly lower numbers of T cells, both total counts and as a proportion of live cells, over the course of the disease than female patients (Extended Data Table 4). Next, we found higher populations of monocytes in both sexes in cohort A (Fig. 2b, c, Extended Data Fig. 2b) compared to HCWs. Although CD14+CD16− classical monocytes were comparable across all groups, levels of CD14+CD16+ intermediate monocytes were increased in patients compared with HCWs, and this increase was more robust in female patients (Fig. 2b, c). By contrast, male patients had higher levels of CD14loCD16+ non-classical monocytes than controls and female patients (Fig. 2b, c). These differences were observed in age- and BMI-adjusted analyses, too, but were not significant (Extended Data Table 3).
We then divided the 17 cohort A male patients into two groups, namely, a ‘high’ group who had high percentages of non-classical monocytes (upper quartile 4 patients, all had more than 5% of non-classical monocytes) and a ‘low-intermediate’ group (others, 13 patients). We compared age, BMI, DFSO, T cells, and plasma levels of IL-18 and CCL5. Although we found no differences in age, BMI or DFSO (Fig. 2d), we noted that the group with high levels of non-classical monocytes had significantly lower levels of T cells and higher levels of CCL5 in plasma (Fig. 2d). In addition, we found a significant correlation between CCL5 levels and abundance in non-classical monocytes only in male patients (Fig. 2e). These findings suggest that the progression from classical to non-classical monocytes may be arrested at the intermediate stage in female patients, and that increased innate inflammatory cytokines and chemokines are associated with more robust activation of innate immune cells at the baseline as well as more robust longitudinal T cell decrease in male patients.
Higher T cell activation in female
We further examined the T cell phenotype in patients with COVID-19. The composition of overall CD4-positive and CD8-positive cells among T cells were similar between all groups in cohort A (Fig. 3a, Extended Data Fig. 2c, Extended Data Table 3). Detailed phenotyping of T cells for naive T cells, central or effector memory T (TCM/TEM) cells, follicular helper T (TFH) cells, regulatory T (Treg) cells revealed no remarkable differences in the frequency of these subsets between sexes (Extended Data Fig. 2c). However, we observed higher levels of CD38 and HLA-DR-positive activated T cells in female patients than in male patients (Fig. 3b, c). In parallel, PD-1- and TIM-3-positive terminally differentiated T cells were more prevalent among female patients than male patients (Fig. 3d, e). These findings were seen in both CD4 and CD8 T cells, but the differences were more robust in CD8 T cells (Fig. 3c, e, Extended Data Table 3). We also stained for intracellular cytokines such as IFNγ, granzyme B (GzB), TNF, IL-6 and IL-2 in CD8 T cells, and IFNγ, TNF, IL-17A, IL-6 and IL-2 in CD4 T cells. Levels of these cytokines were higher in patients than in controls, and were generally comparable between sexes in the patients (Extended Data Fig. 2d). Analyses of T cell phenotypes in cohort B did not reveal any significant differences between sexes (Extended Data Tables 4 and 5). Therefore, female patients with COVID-19 had more abundant activated and terminally differentiated T cell populations than male patients at baseline in unadjusted analyses.
Sex-dependent immunity and disease course
We investigated whether certain immune phenotypes were correlated with disease trajectory, and whether these phenotypes and factors differed between the sexes. To this end, we evaluated the disease course of patients in cohort A. The clinical scores at the first sample collection (_C_1) were 1 or 2 for all of the patients in cohort A. The patients were categorized into a ‘deteriorated’ group if the patients marked a score of 3 or higher after the first sample collection date as their maximum clinical scores during admission (_C_max). By contrast, if the patients maintained the score of 1 or 2, they were categorized as ‘stabilized’ (Extended Data Table 2). Both in male (n = 17) and female (n = 22) patients from cohort A, 6 patients of each sex deteriorated during the course of the disease (35.3% and 27.3%, respectively), and the intervals between the dates at which the patients reached _C_max (DFSO at _C_max) and the first sample collection (DFSO at _C_1) were not significantly different between deteriorated male and female patients (mean ± s.d. = 3.7 ± 4.1 and 4.2 ± 2.7, respectively; P = 0.81 by unpaired two-tailed _t_-test).
We first examined age, BMI, viral loads and titres of anti-S1-IgG antibodies between the stabilized and deteriorated groups in a sex-aggregated manner. We found that the deteriorated group had on average a higher BMI than the stabilized group. Although the age was not statistically different, the stabilized group spanned a larger age range than the deteriorated group, who were generally of a more advanced age. The viral load and antibody titres were comparable (Fig. 4a). Next, we examined these factors in a sex-disaggregated manner, and found that the deteriorated male (M_deteriorated) group was on average significantly older than the stabilized male (M_stabilized) group, whereas the two female groups (F_deteriorated and F_stabilized) were comparable in age (Fig. 4b). In addition, BMI was higher for the M_deteriorated than the M_stabilized group, whereas there was no difference in BMI between the F_deteriorated and F_stabilized groups (Fig. 4b). By contrast, the F_deteriorated group had higher viral load in saliva than the F_stabilized group, whereas there was no difference in the male groups (Fig. 4b). The levels of antibodies were comparable between the deteriorated and stabilized groups both in male and female, but stabilized female tended to have higher antibody levels (Fig. 4b).
We further investigated whether the key factors identified in the previous analyses correlated with disease progression in male and female patients. We observed that regardless of sex, some chemokines and growth factors, such as CXCL10 (also known as IP-10) and M-CSF, were increased in patients that went on to develop worse disease. However, there were some innate immune factors, such as CCL5, TNFSF10 (also known as TRAIL) and IL-15, that were specifically increased only in female patients that subsequently progressed to worse disease, but this difference was not observed in male patients (Fig. 4c). In the age- and DFSO-adjusted analysis of cohort A, we also found that CCL5 was only increased in female patients that progressed to worse disease compared to the stabilized patients, but no such correlation was found in male patients (Extended Data Table 6).
T cell phenotypes in these groups showed that male patients whose disease worsened had a significantly lower proportion of activated T cells (CD38+HLA-DR+) and terminally differentiated T cells (PD-1+TIM-3+) and tendencies for fewer IFNγ+ CD8 T cells at the first sample collection, compared with their counterpart male who did not progressed to worse disease (Fig. 4d). However, in female patients, the deteriorated group had similar levels of these types of CD8 T cells compared with the stabilized group (Fig. 4d).
We finally examined the correlations between age, BMI, viral loads, anti-S1 antibodies, cytokines or chemokines, activated or terminally differentiated or IFNγ-producing CD8 T cells, and clinical disease course (‘_C_max − _C_1’ was used for the deterioration score). The correlation matrix showed that in female patients, higher levels of innate immunity cytokines, such as TNFSF10 and IL-15, were positively correlated with disease progression, whereas there was no association between CD8 T cell status and deterioration (Fig. 4e, results of age- and DFSO-adjusted analysis in Extended Data Table 6). In particular, CXCL10, M-CSF and IL-15 were positively correlated with IFNγ+CD8 T cells in female patients (Fig. 4d).
By contrast, in male patients, progressive disease was associated with higher age, higher BMI, and poor CD8 T cell activation (Fig. 4e). Poor CD8 T cell activation and poor production of IFNγ by CD8 T cells were significantly correlated with patients’ age, whereas these correlations were not seen in female patients (Fig. 4e, f). These differences seemed to highlight the differences between the sexes in the immune responses against SARS-CoV-2 as well as the difference of the potential prognostic or predictive factors for clinical deterioration of COVID-19.
Discussion
Our results revealed key differences in immune responses during the disease course of SARS-CoV-2 infection in male and female patients. First, we found that the levels of several important pro-inflammatory innate immunity chemokines and cytokines such as IL-8, IL-18 (at baseline) and CCL5 (longitudinal analysis) were higher in male patients, which correlated with higher non-classical monocytes (at baseline). Second, we observed a more robust T cell response among female patients than male patients at baseline. In particular, activated CD8 T cells were significantly increased only in female patients but not in male patients compared with healthy volunteers. Analysis of their clinical trajectory showed that, although poor T cell responses were associated with future progression of disease in male patients, higher levels of innate immune cytokines were associated with worsening of COVID-19 disease in female patients. Notably, the T cell response was significantly and negatively correlated with patients’ age in male, but not female, patients. These data indicate key differences in the baseline immune capabilities in male and female patients during the early phase of SARS-CoV-2 infection, and suggest a potential immunological underpinning of the distinct mechanisms of disease progression between sexes. These analyses also provide a potential basis for taking sex-dependent approaches to prognosis, prevention, care, and therapy for patient with COVID-19.
Although our study provides a strong basis for further investigation into how COVID-19 disease dynamics may differ between male and female patients, it is important to note that there are some limitations to the analyses presented in this Article. First, we acknowledge that the healthy HCWs used as the control population were not matched to patients on the basis of age, BMI or underlying risk factors. To account for this, we performed adjusted analyses for the baseline and longitudinal comparisons between patients (cohort A and the full patient population, cohort B) and HCWs, controlling for age and BMI. However, we cannot rule out residual confounding due to underlying risk factors not available for the HCW controls.
Collectively, these data suggest that vaccines and therapies to increase T cell immune responses to SARS-CoV-2 might be warranted for male patients, whereas female patients might benefit from therapies that dampen innate immune activation early during disease. The immune landscape in patients with COVID-19 is considerably different between the sexes, and these differences may underlie heightened disease vulnerability in men.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. Investigators were blinded during experiments in terms of the sex or other clinical background information, with the sample labels having de-identified patient IDs that did not contain any of this information.
Ethics statement
This study was approved by Yale Human Research Protection Program Institutional Review Boards (FWA00002571, Protocol ID. 2000027690). Informed consent was obtained from all enrolled patients and healthcare workers.
Patients and HCWs
Adult patients (≥18 years old) admitted to Yale-New Haven Hospital between 18 March and 9 May 2020, positive for SARS-CoV-2 by RT–PCR from nasopharyngeal and/or oropharyngeal swabs, and able to provide informed consent (surrogate consent accepted) were eligible for the Yale IMPACT Biorepository study, and 198 patients were enrolled in this period. All patients necessitated hospitalization for their symptoms and had an WHO score[17](/articles/s41586-020-2700-3#ref-CR17 "WHO. R&D Blueprint Novel Coronavirus COVID-19 Therapeutic Trial Synopsis https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf
(2020).") of at least 3 at admission (denoting hospitalized, mild disease). At the initial screening, clinical PCR tests were performed in CLIA-certified laboratory and only the PCR-positive patients were enrolled. Only after the confirmation of PCR-positivity, the patients were enrolled and the first time point samples for this study were collected for each patient. The first time point samples were collected at 11.4 ± 8.1, 10.2 ± 6.3, 11.7 ± 7.2 and 12.1 ± 7.3 (mean ± s.d.) DFSO in cohort A female, cohort A male, cohort B female and cohort B male, respectively (Extended Data Fig. [1a](/articles/s41586-020-2700-3#Fig5), right panel for cohort A, Extended Data Table [1](/articles/s41586-020-2700-3#Tab1)).Among these patients, we could obtain whole blood for flow cytometry analysis using fresh PBMCs, plasma for cytokine or chemokine measurements, anti-S1 antibody measurements and nasopharyngeal swab and saliva from total of 98 individuals for the present study. For longitudinal analyses, biospecimens (blood, nasopharyngeal swabs, saliva, urine, and/or stool) were collected at study enrolment (baseline) and on average every 3 to 7 days while in the hospital in 48 of these 98 patients.
The patients were assessed with a locally developed clinical scoring system for disease severity; (1): admitted and observed without supplementary oxygen; (2) required ≤ 3 l supplementary oxygen via nasal canal to maintain SpO2 > 92%; (3) received tocilizumab, which per hospital treatment protocol required that the patient to require >3 l supplementary oxygen to maintain SpO2 > 92%, or, required >2 l supplementary oxygen to maintain SpO2 > 92% and had a high-sensitivity C-reactive protein (CRP) > 70; (4) the patient required ICU-level care; (5) the patient required intubation and mechanical ventilation. In relation to the WHO scoring[17](/articles/s41586-020-2700-3#ref-CR17 "WHO. R&D Blueprint Novel Coronavirus COVID-19 Therapeutic Trial Synopsis https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf
(2020)."), our clinical scores 1, 2/3, 4 and 5 largely correspond to WHO scores 3, 4, 5 and 6/7, respectively. Detailed demographic information for the entire cohort (98 cohort B patients, and several time-point samples from 54 patients among them) and of cohort A (39 patients) are shown in Extended Data Tables [1](/articles/s41586-020-2700-3#Tab1)–[3](/articles/s41586-020-2700-3#Tab3). For the patients who are 90 years old or older, their ages were protected health information, and ‘90’ was put as the surrogate value for the analyses. Among 198 total patients enrolled in IMPACT study in this period, we obtained whole blood, nasopharyngeal swabs or saliva samples from 98 patients for the present study. Individuals with active chemotherapy against cancers, pregnant patients, patients with background haematological abnormalities, patients with autoimmune diseases and patients with a history of organ transplantation and on immunosuppressive agents, were excluded from this study.As a control group, COVID-19-uninfected HCWs from Yale-New Haven Hospital were enrolled. HCWs were tested every 2 weeks for PCR and serology. For the control group, the PBMCs and plasma analysis were done when both tests were negative. That is, if either or both of these tests were positive, these samples were excluded from the analyses. In some HCWs, samples were collected for the assays at up to two time points. In these cases, if the data for a certain type of assay were available for both of these time points, only the first time point data were used and otherwise data for either time point were used in the main analyses with cohort A.
Viral RNA measurement
SARS-CoV-2 RNA concentrations were measured from nasopharyngeal samples and saliva samples by RT–PCR as previously described18,[19](/articles/s41586-020-2700-3#ref-CR19 "Wyllie, A. L. et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N. Engl. J. Med. https://doi.org/10.1056/NEJMc2016359
(2020)."). In short, total nucleic acid was extracted from 300 μl of viral transport media from the nasopharyngeal swab or 300 μl of whole saliva using the MagMAX Viral/Pathogen Nucleic Acid Isolation kit (ThermoFisher Scientific) using a modified protocol and eluted into 75 μl of elution buffer[19](/articles/s41586-020-2700-3#ref-CR19 "Wyllie, A. L. et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N. Engl. J. Med.
https://doi.org/10.1056/NEJMc2016359
(2020)."). For SARS-CoV-2 RNA detection, 5 μl of RNA template was tested as previously described[18](/articles/s41586-020-2700-3#ref-CR18 "Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 5, 1299–1305 (2020)"), using the US CDC real-time RT–PCR primer/probe sets for 2019-nCoV\_N1, 2019-nCoV\_N2, and the human RNase P (RP) as an extraction control. Virus RNA copies were quantified using a tenfold dilution standard curve of RNA transcripts that we previously generated[18](/articles/s41586-020-2700-3#ref-CR18 "Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 5, 1299–1305 (2020)"). If the RNA concentration was lower than the limit of detection (ND) that was determined previously[18](/articles/s41586-020-2700-3#ref-CR18 "Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 5, 1299–1305 (2020)"), the value was set to 0 and used for the analyses.Isolation of plasma
Plasma samples were collected after whole blood centrifugation at 400_g_ for 10 min at room temperature with brake off. The plasma was then carefully transferred to 15-ml conical tubes and then aliquoted and stored at −80 °C for subsequent analysis.
SARS-CoV-2 specific antibody measurement
ELISAs were performed as previously described20. In short, Triton X-100 and RNase A were added to serum samples at final concentrations of 0.5% and 0.5 mg ml−1 respectively and incubated at room temperature for 30 min before use to reduce risk from any potential virus in serum. Then, 96-well MaxiSorp plates (Thermo Scientific 442404) were coated with 50 μl per well of recombinant SARS-CoV-2 S1 protein (ACROBiosystems S1N-C52H3-100 μg) at a concentration of 2 μg ml−1 in PBS and were incubated overnight at 4 °C. The coating buffer was removed, and plates were incubated for 1 h at room temperature with 200 μl of blocking solution (PBS with 0.1% Tween-20, 3% milk powder). Serum was diluted 1:50 in dilution solution (PBS with 0.1% Tween-20, 1% milk powder) and 100 μl of diluted serum was added for two hours at room temperature. Plates were washed three times with PBS-T (PBS with 0.1% Tween-20) and 50 μl of HRP anti-Human IgG Antibody (GenScript A00166, 1:5,000) or anti-Human IgM-Peroxidase Antibody (Sigma-Aldrich A6907, 1:5,000) diluted in dilution solution were added to each well. After 1 h of incubation at room temperature, plates were washed six times with PBS-T. Plates were developed with 100 μl of TMB Substrate Reagent Set (BD Biosciences 555214) and the reaction was stopped after 12 min by the addition of 100ul of 2 N sulfuric acid. Plates were then read at a wavelength of 450 nm and 570 nm.
The cut-off values for sero-positivity were determined as 0.392 and 0.436 for anti-S1-IgG and anti-S1 IgM, respectively. Eighty pre-pandemic plasma samples were assayed to establish the negative baselines, and these values were statistically determined with confidence level of 99%.
Cytokine and chemokine measurement
Patients’ sera isolated as above were stored in −80 °C until the measurement of the cytokines. The sera were shipped to Eve Technologies on dry ice, and levels of 71 cytokines and chemokines were measured with Human Cytokine Array/Chemokine Array 71-Plex Panel (HD71). All the samples were measured upon the first thaw.
The shipment of the samples and measurements were done in two separate batches, but the measurements were performed with the same assay kits using the same standard curves, therefore minimizing the batch effects between the measurements.
For the out of range values of the measurements, either the lowest highest extrapolatable values or the lowest or highest standard curve were recorded following the instructions of HD71 assay, and included in the analyses. Among all the samples measured, we found that two samples had outlier values (beyond 1.5× interquartile range) in more than half of the 71 cytokines or chemokines measured, suggesting the technical error and/or poor sample qualities in the measurements. Therefore, cytokine or chemokine data of these individuals were excluded from the analyses.
Isolation of PBMCs
The PBMCs were isolated from heparinized whole blood using Histopaque density gradient under the biosafety level 2+ facility. To isolate PBMCs, blood 1:1 diluted in PBS was layered over in Histopaque in a SepMate tube and centrifuged for 10 min at 1,200_g_. The PBMC layer was collected by quickly pouring the content into a new 50-ml tube. The cells were washed twice with PBS to remove any remaining histopaque and to remove platelets. The pelleted cells were treated with ACK buffer for red cell lysis and then counted. The percentage viability was estimated using Trypan blue staining.
Flow cytometry
Using the freshly isolated PBMCs, the staining was performed in three separate panels for (1) PBMC cell composition, (2) T cell surface staining, and (3) T cell intracellular staining. Exact antibody clones and vendors that were used for flow cytometric analysis are as follows: BB515 anti-HLA-DR (G46-6), BV785 anti-CD16 (3G8), PE-Cy7 anti-CD14 (HCD14), BV605 anti-CD3 (UCHT1), BV711 anti-CD19 (SJ25C1), BV421 anti-CD11c (3.9), AlexaFluor647 anti-CD1c (L161), Biotin anti-CD141 (M80), PE anti-CD304 (12C2), APCFire750 anti-CD11b (ICRF44), PerCP/Cy5.5 anti-CD66b (G10F5), BV785 anti-CD4 (SK3), APCFire750 or PE-Cy7 or BV711 anti-CD8 (SK1), BV421 anti-CCR7 (G043H7), AlexaFluor 700 anti-CD45RA (HI100), PE anti-PD1 (EH12.2H7), APC anti-TIM-3 (F38-2E2), BV711 anti-CD38 (HIT2), BB700 anti-CXCR5 (RF8B2), PE-Cy7 anti-CD127 (HIL-7R-M21), PE-CF594 anti-CD25 (BC96), BV711 anti-CD127 (HIL-7R-M21), BV421 anti-IL-17a (N49-653), AlexaFluor 700 anti-TNF (MAb11), PE or APC/Fire750 anti-IFNy (4S.B3), FITC anti-GranzymeB (GB11), AlexaFluor 647 anti-IL-4 (8D4-8), BB700 anti-CD183/CXCR3 (1C6/CXCR3), PE-Cy7 anti-IL-6 (MQ2-13A5), PE anti-IL-2 (5344.111), BV785 anti-CD19 (SJ25C1), BV421 anti-CD138 (MI15), AlexaFluor700 anti-CD20 (2H7), AlexaFluor 647 anti-CD27 (M-T271), PE/Dazzle594 anti-IgD (IA6-2), PE-Cy7 anti-CD86 (IT2.2), APC/Fire750 anti-IgM (MHM-88), BV605 anti-CD24 (M1/69), APC/Fire 750 anti-CD10 (HI10a), BV421 anti-CD15 (SSEA-1), AlexaFluor 700 Streptavidin (ThermoFisher). Freshly isolated PBMCs were plated at 1 × 106–2 × 106 cells in a 96-well U-bottom plate. Cells were resuspended in Live/Dead Fixable Aqua (ThermoFisher) for 20 min at 4 °C. Following a wash, cells were then blocked with Human TruStan FcX (BioLegend) for 10 min at room temperature. Cocktails of desired staining antibodies were directly added to this mixture for 30 min at room temperature. For secondary stains, cells were washed and supernatant aspirated; to each cell pellet, a cocktail of secondary markers was added for 30 min at 4 °C. Before analysis, cells were washed and resuspended in 100 μl of 4% paraformaldehyde for 30 min at 4 °C. For intracellular cytokine staining following stimulation, cells were resuspended in 200 μl cRPMI (RPMI-1640 supplemented with 10% FBS, 2 mM l-glutamine, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol) and stored at 4 °C overnight. Subsequently, these cells were washed and stimulated with 1× Cell Stimulation Cocktail (eBioscience) in 200 μl cRPMI for 1 h at 37 °C. Directly to this, 50 μl of 5× Stimulation Cocktail (plus protein transport inhibitor) (eBioscience) was added for an additional 4 h of incubation at 37 °C. After stimulation, cells were washed and resuspended in 100 μl of 4% paraformaldehyde for 30 min at 4 °C. To quantify intracellular cytokines, these samples were permeabilized with 1× Permeabilization Buffer from the FOXP3/Transcription Factor Staining Buffer Set (eBioscience) for 10 min at 4 °C. All further staining cocktails were made in this buffer. Permeabilized cells were then washed and resuspended in a cocktail containing Human TruStan FcX (BioLegend) for 10 min at 4 °C. Finally, intracellular staining cocktails were directly added to each sample for 1 h at 4 °C. After this incubation, cells were washed and prepared for analysis on an Attune NXT (ThermoFisher). Data were analysed using FlowJo software v.10.6 software (Tree Star).
Set of markers used to identify each subset of cells are summarized in Extended Data Table 7, and gating strategies for the key cell populations presented in the main figures are shown in Extended Data Fig. 3a–c. For most samples, all available staining panels were implemented and analysed. The few exceptions pertained to those samples during which a mechanical malfunction occurred, which depleted the sample before acquisition, or to the samples with poor staining qualities. In these cases, data for these samples or panels were missing and not available. All the data available were used for the analyses, and the data used to generate figures and tables can be found in Supplementary Table 1, and the raw fcs files are available at ImmPort as described in the 'Data Availability' section.
Statistical analysis for the primary analyses
For the primary analyses shown in the main figures, Graph Pad Prism (v,8.0) was used for all statistical analysis. Unless otherwise noted, one-way ANOVA with Bonferroni’s multiple comparison test was used for the comparisons between M_Pt versus F_Pt, M_Pt versus M_HCW, F_Pt versus F_HCW, and M_HCW versus F_HCW for the comparisons. For two-group comparisons including the comparison between stabilized group and deteriorated group in each sex (Fig. 4a–d), two-sided unpaired _t_-test was used for the comparison. Bioconductor R (v.3.6.3) package ggplot2 (v.3.3.0) was used to generate heat maps (Extended Data Fig. 2), X_–_Y graphs for correlation analyses (Figs. 2e, 4f), and Pearson correlation heat maps (Fig. 4e).
Statistical analysis for the secondary analyses
All multivariable analyses were conducted using R v.3.6.1 (for data cleaning) and SAS version 9.4 (Cary, NC; for data analysis). The code used for data cleaning and data analysis is available at https://github.com/muhellingson/covid_immresp. We conducted longitudinal analyses of the differences in immune response by sex for patients with COVID-19 and differences in immune response between patients with COVID-19 and HCWs by sex and adjusted linear regression to evaluate differences in immune response by sex at baseline and the differences in immune response by sex and patient trajectory.
Longitudinal difference in immune response in all patients positive for COVID-19 (cohort B) by sex
A marginal linear model was fitted to evaluate the difference in various immune responses (outcome) in patients by sex (exposure). We used an auto-regressive correlation structure to account for correlation between repeated observations in an individual over time. To account for the small sample size and unequal follow-up between participants, we used the Morel–Bokossa–Neerchal (MBN) correction. In addition to sex, the model contained time-independent terms for age (in years) and BMI and time-dependent terms for days from symptom onset (self-reported), ICU status (as a proxy for disease severity) and treatment with either tocilizumab or corticosteroids. A patient was defined as ‘on tocilizumab’ at a given time point if they had received the treatment within 14 days before the time the sample was taken. Patients were defined as ‘on corticosteroids’ if they had received the treatment on the same day the sample was taken. The resulting regression coefficients were interpreted as the difference in the adjusted least square means immune response between female and male patients.
Difference in immune response between patients with COVID-19 (cohort A) and HCWs by sex at baseline
We used linear regression to evaluate the difference in immune response between female and male patients at the first time point for those patients who had not received corticosteroids or tocilizumab before enrolment (cohort A). The model contained terms for sex, patient trajectory (worsened versus stable), age, BMI, and an interaction term for sex and group (patient versus HCWs). We calculated the least square means for each group (female patients who worsened, female patients who stabilized, male patients who worsened and male patients who stabilized) and evaluated the differences in the least square means of the different immune response outcomes by group and sex. P values and 95% confidence intervals were calculated with a Tukey correction for multiple pairwise comparisons. The regression coefficient of the interaction term between sex and group was interpreted as the difference-in-differences of the two comparisons by sex or by group (for example, the difference-in-differences between female and male patients and female and male HCWs).
Longitudinal difference in immune response between all patients with COVID-19 (cohort B) and HCWs by sex
We used a marginal linear model with a compound symmetric correlation structure and the MBN correction to evaluate the difference in immune responses between patients and HCWs by sex, controlling for age and BMI. We calculated the least square means for each group (female patients, female HCWs, male patient, male HCWs) and evaluated the differences in adjusted least square means to compare study groups by sex (female patients versus male patients, female HCWs versus male HCWs, female patients versus female HCWs and male patients versus male HCWs). P values and 95% confidence intervals were corrected using the Tukey correction for multiple pairwise comparisons. The regression coefficient of the interaction term between sex and study group was interpreted as the difference-in-differences between the two comparisons by sex or by group.
Multivariable patient trajectory analysis
We used linear regression to evaluate the difference in baseline immune response between patients who worsened after the baseline sample was taken and those who stabilized by sex. The model contained terms for sex, patient trajectory (worsened versus stable), age, days from symptom onset and an interaction term for sex and patient trajectory. We calculated the adjusted least square means for each group (female patients who worsened, female patients who stabilized, male patients who worsened and male patients who stabilized) and evaluated the differences in least square means of the different immune responses by patient trajectory and sex using the Tukey correction for multiple comparisons. The regression coefficient of the interaction term between sex and patient trajectory was interpreted as the difference-in-differences between the two patient trajectories by sex or sex by the two patient trajectories.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All of the background information of HCWs, clinical information of patients, and raw data used in this study are included in the Supplementary Table 1. In addition, all of the raw fcs files for the flow cytometry analysis are uploaded in ImmPort (https://www.immport.org/shared/home, study ID: SDY1648).
References
- Chen, N. et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 395, 507–513 (2020).
Article CAS Google Scholar - Li, Q. et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 382, 1199–1207 (2020).
Article CAS Google Scholar - Yang, X. et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 8, 475–481 (2020).
Article CAS Google Scholar - Meng, Y. et al. Sex-specific clinical characteristics and prognosis of coronavirus disease-19 infection in Wuhan, China: a retrospective study of 168 severe patients. PLoS Pathog. 16, e1008520 (2020).
Article Google Scholar - Gebhard, C., Regitz-Zagrosek, V., Neuhauser, H. K., Morgan, R. & Klein, S. L. Impact of sex and gender on COVID-19 outcomes in Europe. Biol. Sex Differ. 11, 29 (2020).
Article CAS Google Scholar - Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Article CAS Google Scholar - WHO. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19 - 11 March 2020 https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (2020).
- Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020).
Article CAS Google Scholar - Klein, S. L. & Flanagan, K. L. Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638 (2016).
Article CAS Google Scholar - Fischer, J., Jung, N., Robinson, N. & Lehmann, C. Sex differences in immune responses to infectious diseases. Infection 43, 399–403 (2015).
Article CAS Google Scholar - Guerra-Silveira, F. & Abad-Franch, F. Sex bias in infectious disease epidemiology: patterns and processes. PLoS One 8, e62390 (2013).
Article ADS CAS Google Scholar - Moore, A. L. et al. Virologic, immunologic, and clinical response to highly active antiretroviral therapy: the gender issue revisited. J. Acquir. Immune Defic. Syndr. 32, 452–461 (2003).
Article Google Scholar - Collazos, J., Asensi, V. & Cartón, J. A. Sex differences in the clinical, immunological and virological parameters of HIV-infected patients treated with HAART. AIDS 21, 835–843 (2007).
Article Google Scholar - Fink, A. L., Engle, K., Ursin, R. L., Tang, W. Y. & Klein, S. L. Biological sex affects vaccine efficacy and protection against influenza in mice. Proc. Natl Acad. Sci. USA 115, 12477–12482 (2018).
Article CAS Google Scholar - Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Article CAS Google Scholar - Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
CAS PubMed PubMed Central Google Scholar - WHO. R&D Blueprint Novel Coronavirus COVID-19 Therapeutic Trial Synopsis https://www.who.int/blueprint/priority-diseases/key-action/COVID-19_Treatment_Trial_Design_Master_Protocol_synopsis_Final_18022020.pdf (2020).
- Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 5, 1299–1305 (2020)
Article CAS Google Scholar - Wyllie, A. L. et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N. Engl. J. Med. https://doi.org/10.1056/NEJMc2016359 (2020).
- Amanat, F. et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 26, 1033–1036 (2020).
Article CAS Google Scholar
Acknowledgements
We thank M. Linehan for technical and logistical assistance. This work was supported by the Women’s Health Research at Yale Pilot Project Program (A.I., A.M.R.), Fast Grant from Emergent Ventures at the Mercatus Center, Mathers Foundation, the Beatrice Kleinberg Neuwirth Fund, Yale Institute for Global Health, and the Ludwig Family Foundation. IMPACT received support from the Yale COVID-19 Research Resource Fund. A.I. is an Investigator of the Howard Hughes Medical Institute. CBFV is supported by NWO Rubicon 019.181EN.004. A.M. is supported by NIH grant R37AI041699.
Author information
Author notes
- These authors contributed equally: Takehiro Takahashi, Mallory K. Ellingson, Patrick Wong, Benjamin Israelow, Carolina Lucas, Jon Klein, Julio Silva, Tianyang Mao
Authors and Affiliations
- Department of Immunobiology, Yale University School of Medicine, New Haven, CT, USA
Takehiro Takahashi, Patrick Wong, Benjamin Israelow, Carolina Lucas, Jon Klein, Julio Silva, Tianyang Mao, Ji Eun Oh, Maria Tokuyama, Peiwen Lu, Arvind Venkataraman, Annsea Park, Feimei Liu, Eric Y. Wang, Molly L. Bucklin, Yiyun Cao, William Khoury-Hanold, Daniel Kim, Eriko Kudo, Melissa Linehan, Alice Lu-Culligan, Anjelica Martin, Tyler Rice, Eric Song, Nicole Sonnert, Yexin Yang, Aaron M. Ring & Akiko Iwasaki - Department of Epidemiology of Microbial Diseases, Yale School of Public Health, New Haven, CT, USA
Mallory K. Ellingson, Arnau Casanovas-Massana, Anne L. Wyllie, Chantal B. F. Vogels, Rebecca Earnest, Sarah Lapidus, Isabel M. Ott, Adam J. Moore, Kristina Brower, Chaney Kalinich, M. Catherine Muenker, Isabel Ott, Mary Petrone, Annie Watkins, Elizabeth B. White, Nathan D. Grubaugh, Albert I. Ko & Saad B. Omer - Department of Medicine, Section of Infectious Diseases, Yale University School of Medicine, New Haven, CT, USA
Benjamin Israelow, Melissa Campbell, Rupak Datta, Subhasis Mohanty, Haowei Wang, Albert Shaw, John B. Fournier, Camila D. Odio, Shelli Farhadian & Saad B. Omer - Department of Biomedical Engineering, Yale School of Engineering & Applied Science, New Haven, CT, USA
Feimei Liu - Boyer Center for Molecular Medicine, Department of Microbial Pathogenesis, Yale University, New Haven, CT, USA
Amit Meir - Department of Comparative Medicine, Yale University School of Medicine, New Haven, CT, USA
Jonathan Sun - Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT, USA
Isabel M. Ott - Department of Medicine, Section of Pulmonary and Critical Care Medicine, Yale University School of Medicine, New Haven, CT, USA
Giuseppe DeIuliis, Lokesh Sharma & Charles Dela Cruz - Department of Laboratory Medicine, Yale University School of Medicine, New Haven, CT, USA
Wade L. Schulz - Center for Outcomes Research and Evaluation, Yale-New Haven Hospital, New Haven, CT, USA
Wade L. Schulz - Yale Institute for Global Health, Yale University, New Haven, CT, USA
Amyn A. Malik & Saad B. Omer - Yale School of Nursing, Yale University, Orange, CT, USA
Saad B. Omer - Howard Hughes Medical Institute, Chevy Chase, MD, USA
Akiko Iwasaki - Yale Center for Clinical Investigation, Yale University School of Medicine, New Haven, CT, USA
Kelly Anastasio, Staci Cahill & Maxine Kuang - Department of Neurology, Yale University School of Medicine, New Haven, CT, USA
Michael H. Askenase & Sofia Velazquez - Yale School of Medicine, New Haven, CT, USA
Maria Batsu, Hannah Beatty, Santos Bermejo, Bertie Geng, Laura Glick, Ryan Handoko, Lynda Knaggs, Irene Matos, David McDonald, Maksym Minasyan, Nida Naushad, Allison Nelson, Jessica Nouws, Abeer Obaid, Hong-Jai Park, Xiaohua Peng, Harold Rahming, Kadi-Ann Rose, Lorenzo Sewanan, Denise Shepard, Erin Silva, Michael Simonov, Mikhail Smolgovsky, Yvette Strong, Codruta Todeasa, Jordan Valdez & Pavithra Vijayakumar - Department of Biochemistry and of Molecular Biology, Yale University School of Medicine, New Haven, CT, USA
Sean Bickerton & Edward Courchaine - Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, CT, USA
Joseph Lim - Equity Research and Innovation Center, Yale University, New Haven, CT, USA
Marcella Nunez-Smith - Department of Molecular, Cellular and Developmental Biology, Yale University School of Medicine, New Haven, CT, USA
Sarah Prophet
Authors
- Takehiro Takahashi
You can also search for this author inPubMed Google Scholar - Mallory K. Ellingson
You can also search for this author inPubMed Google Scholar - Patrick Wong
You can also search for this author inPubMed Google Scholar - Benjamin Israelow
You can also search for this author inPubMed Google Scholar - Carolina Lucas
You can also search for this author inPubMed Google Scholar - Jon Klein
You can also search for this author inPubMed Google Scholar - Julio Silva
You can also search for this author inPubMed Google Scholar - Tianyang Mao
You can also search for this author inPubMed Google Scholar - Ji Eun Oh
You can also search for this author inPubMed Google Scholar - Maria Tokuyama
You can also search for this author inPubMed Google Scholar - Peiwen Lu
You can also search for this author inPubMed Google Scholar - Arvind Venkataraman
You can also search for this author inPubMed Google Scholar - Annsea Park
You can also search for this author inPubMed Google Scholar - Feimei Liu
You can also search for this author inPubMed Google Scholar - Amit Meir
You can also search for this author inPubMed Google Scholar - Jonathan Sun
You can also search for this author inPubMed Google Scholar - Eric Y. Wang
You can also search for this author inPubMed Google Scholar - Arnau Casanovas-Massana
You can also search for this author inPubMed Google Scholar - Anne L. Wyllie
You can also search for this author inPubMed Google Scholar - Chantal B. F. Vogels
You can also search for this author inPubMed Google Scholar - Rebecca Earnest
You can also search for this author inPubMed Google Scholar - Sarah Lapidus
You can also search for this author inPubMed Google Scholar - Isabel M. Ott
You can also search for this author inPubMed Google Scholar - Adam J. Moore
You can also search for this author inPubMed Google Scholar - Albert Shaw
You can also search for this author inPubMed Google Scholar - John B. Fournier
You can also search for this author inPubMed Google Scholar - Camila D. Odio
You can also search for this author inPubMed Google Scholar - Shelli Farhadian
You can also search for this author inPubMed Google Scholar - Charles Dela Cruz
You can also search for this author inPubMed Google Scholar - Nathan D. Grubaugh
You can also search for this author inPubMed Google Scholar - Wade L. Schulz
You can also search for this author inPubMed Google Scholar - Aaron M. Ring
You can also search for this author inPubMed Google Scholar - Albert I. Ko
You can also search for this author inPubMed Google Scholar - Saad B. Omer
You can also search for this author inPubMed Google Scholar - Akiko Iwasaki
You can also search for this author inPubMed Google Scholar
Consortia
Yale IMPACT Research Team
- Kelly Anastasio
- , Michael H. Askenase
- , Maria Batsu
- , Hannah Beatty
- , Santos Bermejo
- , Sean Bickerton
- , Kristina Brower
- , Molly L. Bucklin
- , Staci Cahill
- , Melissa Campbell
- , Yiyun Cao
- , Edward Courchaine
- , Rupak Datta
- , Giuseppe DeIuliis
- , Bertie Geng
- , Laura Glick
- , Ryan Handoko
- , Chaney Kalinich
- , William Khoury-Hanold
- , Daniel Kim
- , Lynda Knaggs
- , Maxine Kuang
- , Eriko Kudo
- , Joseph Lim
- , Melissa Linehan
- , Alice Lu-Culligan
- , Amyn A. Malik
- , Anjelica Martin
- , Irene Matos
- , David McDonald
- , Maksym Minasyan
- , Subhasis Mohanty
- , M. Catherine Muenker
- , Nida Naushad
- , Allison Nelson
- , Jessica Nouws
- , Marcella Nunez-Smith
- , Abeer Obaid
- , Isabel Ott
- , Hong-Jai Park
- , Xiaohua Peng
- , Mary Petrone
- , Sarah Prophet
- , Harold Rahming
- , Tyler Rice
- , Kadi-Ann Rose
- , Lorenzo Sewanan
- , Lokesh Sharma
- , Denise Shepard
- , Erin Silva
- , Michael Simonov
- , Mikhail Smolgovsky
- , Eric Song
- , Nicole Sonnert
- , Yvette Strong
- , Codruta Todeasa
- , Jordan Valdez
- , Sofia Velazquez
- , Pavithra Vijayakumar
- , Haowei Wang
- , Annie Watkins
- , Elizabeth B. White
- & Yexin Yang
Contributions
A.I., S.B.O. and A.I.K. conceived the study. C.L., P.W., J.K., J. Silva, T.M. and J.E.O. defined parameters for flow cytometry experiments, collected and processed patient PBMC samples. P.W. acquired and analysed the flow cytometry data. B.I., J.K., C.L. and C.D.O. collected epidemiological and clinical data. F.L., A.M., J. Sun, E.Y.W. and A.M.R. acquired and analysed ELISA data. A.L.W., C.B.F.V., I.M.O., R.E., S.L., P.L., A.V., A.P. and M.T. performed the virus RNA concentration assays. N.D.G. supervised viral RNA concentration assays. A.C.-M. and A.J.M. processed and stored patient specimens, J.B.F., C.D.C. and S.F. assisted in patient recruitment, W.L.S. supervised clinical data management, A.S. coordinated and secured funding for PBMC collection. T.T. designed the analysis scheme, analysed and interpreted the data for the baseline analyses. M.K.E. and S.B.O. designed the analysis scheme, and interpreted the data for the longitudinal analyses. M.K.E. analysed the longitudinal data. T.T., M.K.E. and A.I. drafted the manuscript. A.I., A.M.R. and S.B.O. revised the manuscript. A.I. secured funds and supervised the project. Authors from the Yale IMPACT Research Team contributed to collection and storage of patient samples, as well as the collection of the patients’ epidemiological and clinical data.
Corresponding author
Correspondence toAkiko Iwasaki.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Petter Brodin, Malik Peiris and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Comparison of basic clinical parameters of cohort A patient samples and plasma levels of 71 cytokines and chemokines at the first sampling of cohort A.
a, Comparisons of age, BMI and DFSO at the first sampling between male and female patients in cohort A. n = 17 and 22 for M_Pt and F_Pt, respectively. b, Comparison of the plasma levels of 71 cytokines and chemokines. n = 15, 28, 16 and 19 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. Data are mean ± s.e.m. P values were determined by unpaired two-tailed _t_-test (a) or one-way ANOVA with Bonferroni multiple comparison test (b). All P values < 0.10 are shown.
Extended Data Fig. 2 Heat maps of cytokines and chemokines, PBMC composition, T cell subsets, and T cell cytokine expression at the first sampling of cohort A patients.
a, A heat map of the plasma levels (pg ml−1) of 71 cytokines and chemokines. n = 15, 28, 16 and 19 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. b, A heat map for the composition of PBMCs (percentage in live PBMCs). n = 6, 42, 16 and 21 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. c, A heat map for the T cell subsets (percentage in CD3+ cells). n = 6, 45, 16 and 22 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. d, A heat map for the intracellular cytokine staining of T cells (percentage in CD3+ cells). n = 6, 43, 16 and 22 for M_HCW, F_HCW, M_Pt and F_Pt, respectively. In all of these heat maps, log-transformed values were used for heat map generation.
Extended Data Fig. 3 Flow cytometry gating strategy.
a–c, Gating strategy used for monocytes (a), CD38+HLA-DR+ and PD-1+TIM-3+ CD4 or CD8 T cells (b), and T cell intracellular staining for IFNγ+ CD8 T cells (c).
Extended Data Table 1 Demographic and clinical characteristics of cohort A, cohort B and HCW comparison groups
Extended Data Table 2 Background and sample information of 39 cohort A patients
Extended Data Table 3 Adjusted least square means difference in immune response at baseline between male and female patients with COVID-19 in cohort A and male and female HCW controls
Extended Data Table 4 Adjusted least square means difference over time in immune response between male and female patients with COVID-19 in cohort B
Extended Data Table 5 Adjusted least square means difference over time in immune response between male and female patients with COVID-19 in cohort B and male and female healthy HCW controls
Extended Data Table 6 Adjusted least square means difference between male and female patients with COVID-19 in cohort A by patient trajectory
Extended Data Table 7 Definitions of each cell subset in flow cytometry with specific markers
Supplementary information
Rights and permissions
About this article
Cite this article
Takahashi, T., Ellingson, M.K., Wong, P. et al. Sex differences in immune responses that underlie COVID-19 disease outcomes.Nature 588, 315–320 (2020). https://doi.org/10.1038/s41586-020-2700-3
- Received: 04 June 2020
- Accepted: 19 August 2020
- Published: 26 August 2020
- Issue Date: 10 December 2020
- DOI: https://doi.org/10.1038/s41586-020-2700-3