Reactive astrocyte nomenclature, definitions, and future directions (original) (raw)
- Consensus Statement
- Published: 15 February 2021
- Elena Galea ORCID: orcid.org/0000-0003-4537-98972,3 na2,
- András Lakatos ORCID: orcid.org/0000-0002-1301-22924,5 na1,
- James P. O’Callaghan6 na1,
- Gabor C. Petzold ORCID: orcid.org/0000-0002-0145-86417,8 na1,
- Alberto Serrano-Pozo ORCID: orcid.org/0000-0003-0899-75309,10 na1,
- Christian Steinhäuser11 na1,
- Andrea Volterra ORCID: orcid.org/0000-0003-1069-160212 na1,
- Giorgio Carmignoto13,14 na1,
- Amit Agarwal15,
- Nicola J. Allen16,
- Alfonso Araque ORCID: orcid.org/0000-0003-3840-114417,
- Luis Barbeito ORCID: orcid.org/0000-0002-3047-232X18,
- Ari Barzilai19,
- Dwight E. Bergles20,
- Gilles Bonvento ORCID: orcid.org/0000-0002-2886-42281,
- Arthur M. Butt ORCID: orcid.org/0000-0001-7579-074621,
- Wei-Ting Chen22,
- Martine Cohen-Salmon23,
- Colm Cunningham24,
- Benjamin Deneen ORCID: orcid.org/0000-0002-6335-108125,
- Bart De Strooper22,26,
- Blanca Díaz-Castro27,
- Cinthia Farina ORCID: orcid.org/0000-0002-4466-967628,
- Marc Freeman29,
- Vittorio Gallo ORCID: orcid.org/0000-0002-2429-084530,
- James E. Goldman31,
- Steven A. Goldman ORCID: orcid.org/0000-0002-5498-430332,33,
- Magdalena Götz ORCID: orcid.org/0000-0003-1551-920334,35,
- Antonia Gutiérrez ORCID: orcid.org/0000-0002-6264-615236,37,
- Philip G. Haydon ORCID: orcid.org/0000-0001-5698-669838,
- Dieter H. Heiland ORCID: orcid.org/0000-0002-9258-303339,40,
- Elly M. Hol ORCID: orcid.org/0000-0001-5604-260341,
- Matthew G. Holt ORCID: orcid.org/0000-0002-8958-402742,
- Masamitsu Iino43,
- Ksenia V. Kastanenka44,
- Helmut Kettenmann45,
- Baljit S. Khakh46,
- Schuichi Koizumi ORCID: orcid.org/0000-0001-6184-310647,
- C. Justin Lee48,
- Shane A. Liddelow49,
- Brian A. MacVicar ORCID: orcid.org/0000-0003-4596-462350,
- Pierre Magistretti51,52,
- Albee Messing53,
- Anusha Mishra ORCID: orcid.org/0000-0002-3642-504954,
- Anna V. Molofsky ORCID: orcid.org/0000-0002-4709-241155,
- Keith K. Murai56,
- Christopher M. Norris57,
- Seiji Okada ORCID: orcid.org/0000-0002-5107-820958,
- Stéphane H. R. Oliet59,
- João F. Oliveira ORCID: orcid.org/0000-0002-1005-232860,61,62,
- Aude Panatier ORCID: orcid.org/0000-0002-6107-446359,
- Vladimir Parpura63,
- Marcela Pekna ORCID: orcid.org/0000-0003-2734-823764,
- Milos Pekny65,
- Luc Pellerin66,
- Gertrudis Perea ORCID: orcid.org/0000-0001-5924-917567,
- Beatriz G. Pérez-Nievas68,
- Frank W. Pfrieger ORCID: orcid.org/0000-0001-7085-143169,
- Kira E. Poskanzer ORCID: orcid.org/0000-0003-4830-889170,
- Francisco J. Quintana71,
- Richard M. Ransohoff72,
- Miriam Riquelme-Perez1,
- Stefanie Robel73,
- Christine R. Rose ORCID: orcid.org/0000-0002-9684-359274,
- Jeffrey D. Rothstein ORCID: orcid.org/0000-0003-2001-847075,
- Nathalie Rouach ORCID: orcid.org/0000-0002-5574-888X76,
- David H. Rowitch ORCID: orcid.org/0000-0002-0079-00605,
- Alexey Semyanov ORCID: orcid.org/0000-0002-6800-094277,78,
- Swetlana Sirko ORCID: orcid.org/0000-0001-5950-616X79,80,
- Harald Sontheimer ORCID: orcid.org/0000-0002-5843-987181,
- Raymond A. Swanson ORCID: orcid.org/0000-0002-3664-535982,
- Javier Vitorica37,83,
- Ina-Beate Wanner84,
- Levi B. Wood85,
- Jiaqian Wu86,
- Binhai Zheng87,
- Eduardo R. Zimmer88,
- Robert Zorec ORCID: orcid.org/0000-0002-7478-387589,90,
- Michael V. Sofroniew ORCID: orcid.org/0000-0001-6075-017891 na2 &
- …
- Alexei Verkhratsky ORCID: orcid.org/0000-0003-2592-989892,93 na2
Nature Neuroscience volume 24, pages 312–325 (2021)Cite this article
- 96k Accesses
- 1142 Citations
- 324 Altmetric
- Metrics details
Subjects
Abstract
Reactive astrocytes are astrocytes undergoing morphological, molecular, and functional remodeling in response to injury, disease, or infection of the CNS. Although this remodeling was first described over a century ago, uncertainties and controversies remain regarding the contribution of reactive astrocytes to CNS diseases, repair, and aging. It is also unclear whether fixed categories of reactive astrocytes exist and, if so, how to identify them. We point out the shortcomings of binary divisions of reactive astrocytes into good-vs-bad, neurotoxic-vs-neuroprotective or A1-vs-A2. We advocate, instead, that research on reactive astrocytes include assessment of multiple molecular and functional parameters—preferably in vivo—plus multivariate statistics and determination of impact on pathological hallmarks in relevant models. These guidelines may spur the discovery of astrocyte-based biomarkers as well as astrocyte-targeting therapies that abrogate detrimental actions of reactive astrocytes, potentiate their neuro- and glioprotective actions, and restore or augment their homeostatic, modulatory, and defensive functions.
Similar content being viewed by others
Main
‘Neuroglia’ or ‘glia’ are collective terms describing cells of neuroepithelial (oligodendrocytes, astrocytes, oligodendrocyte progenitor cells, ependymal cells), neural crest (peripheral glia), and myeloid (microglia) origin. Changes in neuroglia associated with diseases of the CNS have been noted, characterized, and conceptualized from the very dawn of neuroglial research. Rudolf Virchow, in a lecture to students and medical doctors in 1858, stressed that ‘this very interstitial tissue [that is, neuroglia] of the brain and spinal marrow is one of the most frequent seats of morbid change...’1. Changes in the shape, size, or number of glial cells in various pathological contexts have been frequently described by prominent neuroanatomists2. In particular, hypertrophy of astrocytes was recognized very early as an almost universal sign of CNS pathology: ‘the protoplasmic glia elements [that is, astrocytes] are really the elements which exhibit a morbid hypertrophy in pathological conditions’3. Neuroglial proliferation was thought to accompany CNS lesions, leading to early suggestions that proliferating glia fully replaced damaged neuronal elements4. Thus, a historical consensus was formed that a change in ‘the appearance of neuroglia serves as a delicate indicator of the action of noxious influences upon the central nervous system,’ and the concept of ‘reactionary change or gliosis’ was accepted5. While the origin of ‘gliosis’ is unclear (glia + osis in Greek means ‘glial condition or process’; in Latin the suffix -osis acquired the additional meaning of ‘disease’; hence ‘astrogliosis’ may also carry a connotation of ‘glial disorder’), the term became universally adopted to denote astrocytic remodeling in response to pathologic conditions. The role of reactive astrocytes in forming a scar-border to seal the nervous tissue against penetrating lesions was recognized, with distinct stages being visualised5. In the 21st century, astrocytes are increasingly viewed as having a critical contribution to neurological disorders. Research into the roles of astrocytes in neurology and psychiatry is accelerating and drawing in increasing numbers of researchers. This rapid expansion has exposed a pressing need for unifying nomenclature and refining of concepts6. Here we start by providing a working consensus on nomenclature and definitions and by critically evaluating widely used markers of reactive astrocytes. Then, we describe the advances and take positions on controversies regarding the impact of astrocytes in CNS diseases and aging. Finally, we discuss the need for new names to grasp astrocyte heterogeneity, and we outline a systematic approach to unraveling the contribution of astrocytes to disorders of the CNS. This article is expected to inform clinical thinking and research on astrocytes and to promote the development of astrocyte-based biomarkers and therapies.
Too many names
‘Astrocytosis’, ‘astrogliosis’, ‘reactive gliosis’, ‘astrocyte activation’, ‘astrocyte reactivity’, ‘astrocyte re-activation’, and ‘astrocyte reaction’ have been all used to describe astrocyte responses to abnormal events in the CNS, including neurodegenerative and demyelinating diseases, epilepsy, trauma, ischemia, infection, and cancer. We suggest ‘reactive astrogliosis’ to define the process whereby, in response to pathology, astrocytes engage in molecularly defined programs involving changes in transcriptional regulation, as well as biochemical, morphological, metabolic, and physiological remodeling, which ultimately result in gain of new function(s) or loss or upregulation of homeostatic ones. Although for some researchers, particularly neuropathologists, reactive astrogliosis is invariably associated with irreversible changes such as astrocyte proliferation, scar-border formation, and immune-cell recruitment6, these phenomena mainly occur when there is disruption of the blood–brain barrier (Fig. 1a)7. We also support the term ‘astrocyte reactivity’ as being broadly equivalent to reactive astrogliosis, but emphasizing the capacity of astrocytes to adopt distinct state(s) in response to diverse pathologies. Therefore, ‘reactive astrocytes’, referring to the cells undergoing this remodeling, is an umbrella term encompassing multiple potential states. We define ‘state’ as a transient or long-lasting astrocyte condition characterized by a specific molecular profile, specific functions, and distinct impact on diseases, while ‘phenotype’ is the measurable outcome of that state. Importantly, the changes in astrocytes in response to pathological stimuli are not to be confused with the plasticity of healthy astrocytes, which are constantly being activated by physiological signals in the CNS. For this reason, although transitions from physiology to pathology are progressive and sometimes difficult to define, ‘astrocyte activation’ should be reserved for physiological conditions and not used in pathological contexts, which should be referred to as ‘astrocyte reactivity’.
Fig. 1: Multivariate assessment of reactive astrocytes.
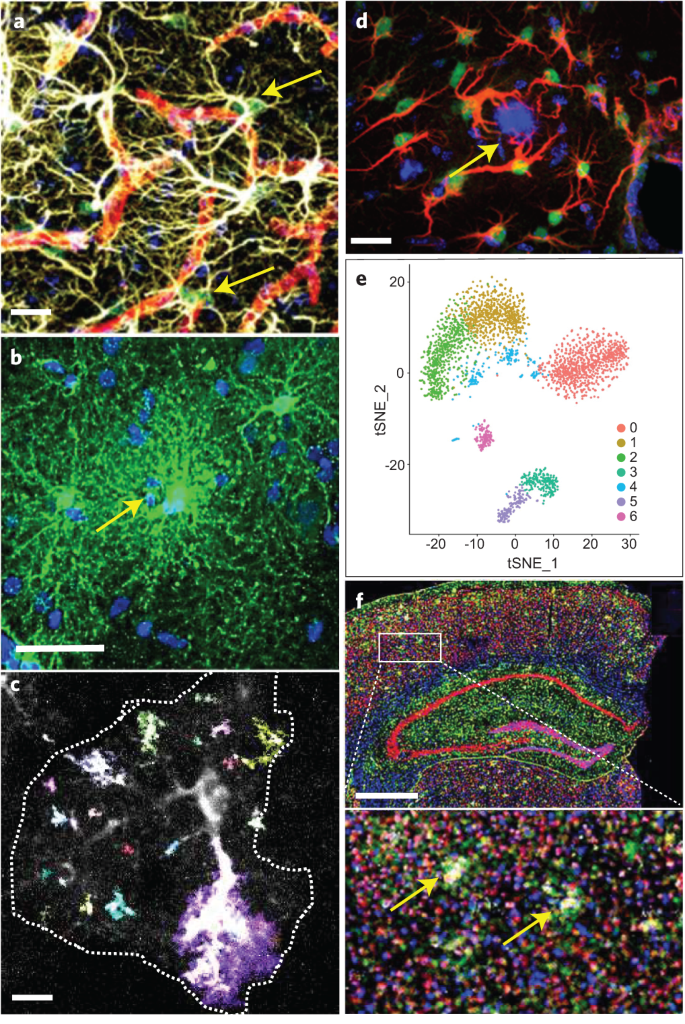
a, Reactive astrocyte proliferation in the vicinity of blood vessels assessed by co-staining for BrdU (green; arrows), DAPI (blue), GFAP (white), and CD31 (red) after stab injury of the mouse cortex. Scale bar, 15 µm. Unpublished image from authors S.S. and M.G. b, Human cortical protoplasmic astrocytes in a surgical specimen injected with Lucifer yellow (arrow, injection site) that traverses the gap junctions into neighboring astrocytes. Scale bar, 45 µm. Courtesy of Drs. Xu, Sosunov, and McKhann, Columbia University Department of Neurosurgery. c, Event-based determination of Ca2+ responses in a GCaMP6-expressing astrocyte (surrounded by a dashed line) in mouse cortical slices using astrocyte quantitative analysis (AQuA)120. Colors indicate AQuA events occurring in a single 1-s frame of a 5-min movie. Scale bar, 10 μm. d, Activation of the transcription factor STAT3 (green) assessed by nuclear accumulation in GFAP+ reactive astrocytes (red) surrounding an amyloid plaque (blue, arrow) in a mouse AD model. Scale bar, 20 µm. Adapted from ref. 121, Society for Neuroscience. e, ScRNAseq in the remission phase of a mouse model of MS reveals several transcriptional astrocyte clusters. These astrocyte sub-populations may be validated with spatial transcriptomics, as shown in f in a model of AD. Adapted from ref. 40, Nature Publishing Group. f, Distribution of 87 astrocytic (green), neuronal (red), microglial (yellow), and oligodendroglial (blue) genes with in situ multiplex gene sequencing in a coronal section from a mouse model of AD. The method ‘reads’ barcodes of antisense DNA probes that simultaneously target numerous mRNAs. Scale bar, 800 μm. Boxed area is magnified in bottom image, showing 6E10+ amyloid-β plaques (white; arrows). Adapted from ref. 96, Cell Press.
The pathological contexts in which astrocyte reactivity occurs can markedly vary and may be sporadic or genetically mediated; acute or chronic; and due to a systemic pathology (for example, sepsis), specific injury or disease of the CNS, or a deleterious experimental manipulation. By definition, astrocyte reactivity is secondary to an extrinsic signal, may evolve with time, and, in many situations, is reversible. Astrocytes may also exhibit cell-autonomous disturbances8, as happens in astrocytopathies resulting from mutated alleles of astrocytic genes (for example, GFAP in Alexander disease)9, as well as from direct viral infections or exposure to toxic substances that specifically damage astrocytes (for example, ammonium in hepatic encephalopathy)10. These astrocytes can be considered ‘diseased astrocytes’ that unequivocally initiate the diseases and may secondarily acquire a reactive phenotype with a distinct impact on disease progression. Mutations in ubiquitously expressed genes, as in familial neurodegenerative disorders (for example, Huntington’s disease, HD), or disease-risk polymorphisms in genes highly expressed in astrocytes (for example, APOE in Alzheimer’s disease, AD)11, may also lead to dysfunctional astrocytes that, without being the sole or primary initiators of pathology, may adversely affect outcomes. Terminology recommendations and caveats are summarized in Box 1 and in the section ‘Are new names needed?’ below.
GFAP as a marker
Glial fibrillary acidic protein (GFAP)—a major protein constituent of astrocyte intermediate filaments—is the most widely used marker of reactive astrocytes (Table 1)12. Indeed, upregulation of GFAP mRNA and protein, as shown with multiple techniques including quantitative PCR (qPCR), RNA sequencing (RNAseq), in situ hybridization, electron microscopy, and immunostaining (Fig. 1a,d), is a prominent feature of many, but not necessarily all, reactive astrocytes: (i) increased GFAP content occurs across diverse types of CNS disorders, (ii) is an early response to injury, and, moreover, (iii) is a sensitive indicator, detectable even in the absence of overt neuronal death (for example, when there is synapse loss, minor demyelination, and extracellular amyloid-β oligomers). However, while the degree of GFAP upregulation in reactive astrocytes often parallels the severity of the injury6, this correlation is not always proportional, perhaps due to regional differences of astrocytes, including basal GFAP content13,14. In the healthy mouse brain, hippocampal astrocytes have a higher GFAP content than cortical, thalamic, or striatal astrocytes; this, however, does not make hippocampal astrocytes more reactive. GFAP is also expressed by progenitor cells15 and its expression depends on developmental stages16,17. In addition, GFAP immunoreactivity has been reported to decrease in a subpopulation of astrocytes in mouse cortex following repetitive trauma6 and in the spinal cord of a mouse model of amyotrophic lateral sclerosis (ALS), probably due to cleavage of GFAP by caspase-3 (ref. 18). Expression of GFAP is also modulated by physiological stimuli such as physical activity19, exposure to enriched environments19, and glucocorticoids20, and it fluctuates with circadian rhythms in the suprachiasmatic nucleus21. Therefore, changes in GFAP expression may also reflect physiological adaptive plasticity rather than being simply a reactive response to pathological stimuli. A common mistake is to interpret higher numbers of GFAP+ cells as local recruitment or proliferation of astrocytes. We recommend using markers of proliferation (Ki67, PCNA, and BrdU incorporation; Table 2) and combining GFAP immunostaining with other ubiquitous astrocyte markers such as aldehyde dehydrogenase-1 L1 (ALDH1L1), glutamine synthetase (GS), and aldolase-C (ALDOC) to correctly estimate astrocyte numbers22, provided that their expression is stable. Finally, there are discrepancies between observed GFAP mRNA and protein levels, perhaps due to differential regulation of translation, post-translational modifications, protein half-life, and antibody epitope accessibility. Overall, although an increase in GFAP content is a strong indication of reactive astrocyte remodeling, it is not an absolute marker of reactivity, nor does it strictly correlate with the extent thereof or indicate altered functions of reactive astrocytes.
Table 1 Potential markers of reactive astrocytes
Table 2 Potential functional assessments for reactive astrocytes
Morphology revisited
Increased GFAP immunoreactivity largely reflects changes in the astrocytic cytoskeleton and tends to exaggerate the degree of hypertrophy because, with the exception of scar-border astrocytes, the volume accessed by reactive astrocytes does not change, as they remain in their territorial domains23. In other words, cytoskeletal reorganization does not necessarily equal astrocyte hypertrophy. Immunohistochemical staining for cytosolic enzymes such as ALDH1L1, ALDOC, GS, and S100B allow the visualization of the somata and proximal processes of astrocytes, although, like GFAP, these markers fail to reveal small processes. Membrane proteins such as the glutamate transporters EAAT1 and EAAT2 are not optimal candidates to assess complex astrocyte morphology, as they tend to produce widespread and diffuse staining24. In addition, the expression of some of these proteins may change in reactive astrocytes22 (Table 1), and some might be expressed by other cell types in specific brain regions13. Animal models expressing fluorescent proteins in the astrocyte cytosol or membrane through astrocyte-specific transgenesis or after gene transfer with viral vectors25 circumvent the limitations of immunohistochemical analysis. Further, dye-filling methods can be used to visualize whole astrocytes in mice23, as well as in human brain samples from surgical resections (Fig. 1b)24. Thorough visualization is necessary because astrocytes undergo distinct morphological changes other than hypertrophy in pathological contexts, including elongation, process extension toward injury site, and some three-dimensional (3D) domain overlap26. In addition, although astrocytes appear to be more resistant than neurons to degeneration and death, loss of primary and secondary astrocyte branches has been reported in mouse models of AD27 and ALS18, and in patients with multiple sclerosis (MS)28. Detailed analyses of astrocyte arborization in CNS diseases and injuries, however, are pending, given that the fine perisynaptic and perivascular astrocytic processes can only be revealed with super-resolution, expansion, or electron microscopy. Finally, clasmatodendrosis (from Greek klasma, fragment, + dendron, tree, + osis, condition or process) is a form of astrodegeneration characterized by an extreme fragmentation or beading and disappearance of distal fine processes, along with swelling and vacuolation of the cell body. It is observed in neuropathological specimens after severe trauma and ischemia, as well as in the aged brain29. However, although astrocytes may suffer plasma membrane disruption due to mechanical damage and cleavage of membrane proteins and cytoskeletal proteins, including GFAP, by proteases in acute brain trauma30,31, the phenomenon of clasmatodendrosis should be approached with caution, because it may be an artifact derived from postmortem autolysis with no pathophysiological bearing, as suggested by Cajal32. In summary, GFAP upregulation and hypertrophy are useful but insufficient markers of astrocyte reactivity that need to be complemented by additional markers (Table 1 and Box 1).
Impact in CNS diseases
Research on astrocytes in CNS diseases has advanced in the last century in line with conceptual and technological progress in astrocyte biology. New approaches have been progressively integrated with existing ones, and these continue to evolve. At present, research in reactive astrocytes is an interdisciplinary endeavor combining -omics approaches with physiology and genetic manipulation. Below, we summarize advances and controversies with regards to the impact of astrocytes in CNS diseases from a historical perspective, punctuated by technical advances.
From morphology to functional studies
From the early 20th century up to the 1980s, the morphological appearance of astrocytes was the only readout of their role in neuropathology. Hypertrophy and increased GFAP content were generally regarded as reflections of a detrimental astrocyte phenotype. The advent of genetic engineering in the early 1990s opened a new phase of research based on astrocyte-targeted manipulation of gene expression. For example, depletion or overexpression of receptors, membrane proteins33,34, cytoskeleton proteins35, acute-phase proteins36, heat-shock proteins37, and transcription factors38,39,40 in astrocytes or ablation of proliferative scar-border forming astrocytes41 was reported to modify (protect or exacerbate) the course of neurological diseases in mouse models. An important conclusion drawn from these studies is that the morphological appearance of astrocytes does not correlate with functional phenotypes or with their impact on other cell types. Moreover, the overall impact of reactive astrocytes on each disease is complex. For example, the manipulation of reactive astrocytes has resulted in improved outcomes38,42,43, worse35 outcomes, and no change44 in mouse models of AD and MS40,45,46. Plausibly, such differences arise from several scenarios: (i) pathways that ultimately exacerbate, attenuate, or have no impact on ongoing pathology occur in the same astrocyte, such that the selective manipulation of one pathway may mask, or secondarily impact, the manifestation of others; (ii) coexisting astrocyte subpopulations may have opposing effects on pathology45; (iii) in neurodegenerative diseases, a spectrum of reactive-astrocyte phenotypes conceivably coexist in the same brain at a given time point because of the asynchronous progression of neuropathology in different brain regions; or (iv) the pathological impact of astrocytes is stage-dependent, as shown in mouse models of MS40,45,46. Finally, pathways inducing astrocyte reactivity may be beneficial in one disease and detrimental in another. For example, activation of STAT3-dependent transcription is beneficial in neonatal white matter injury47, traumatic brain injury30, spinal cord injury48,49, and motor neuron injury50, but detrimental in AD models42,43. That is, STAT3-mediated transcriptional programs may contribute to malfunctional astrocyte states in AD models and to resilient states in other conditions. We broadly define ‘astrocyte resilience’ as the set of successful astroprotective responses that maintain cell-intrinsic homeostatic functions in neural circuits (Table 2) while promoting both neuronal and astrocyte survival. Lastly, responses of reactive astrocytes may be maladaptive and result in malfunctional astrocytes, which, in addition to losing homeostatic functions, may also gain detrimental functions, thus exacerbating ongoing pathology6. Numerous mixed scenarios of malfunctional and resilient astrocytes plausibly exist, with multidirectional transitions among them.
Research in the last decade has begun to unravel specific functional alterations in reactive astrocytes underlying complex phenotypic changes. In normal conditions, astrocyte Ca2+-based responses, and downstream signaling via neuroactive mediators, exert multifarious effects on synaptic function and plasticity, neural-network oscillations, and, ultimately, on behavior51,52. In pathology, various functional changes emerge. Astrocyte Ca2+ dynamics and network responses become aberrant in mouse models of HD53, AD54, and ALS55, possibly contributing to cognitive impairment and neuropathology43,53,56. Reactive microglia may shift astrocyte signaling from physiological to pathological by increasing production of tumor necrosis factor α (TNFα), thus altering synaptic functions and behavior57. Functions lost or altered in reactive astrocytes include neurotransmitter and ion buffering in mouse HD models58, communication via gap junctions in the sclerotic hippocampus of patients with epilepsy59, phagocytic clearance of dystrophic neurites60, and metabolic coupling by glycolysis-derived d-serine61 and lactate62 in mouse AD models. The excessive release of GABA by reactive astrocytes in AD63 and Parkinson’s disease64 may be a case of gain of detrimental function. Another example may be what is sometimes called ‘astrocyte neurotoxicity’, but we recommend using this term only when increased neuronal death is due to the verified release of an identified toxic factor by reactive astrocytes, and not merely due to loss of trophic or antioxidant support from astrocytes. An example is neuronal damage due to nitrosative stress caused by astrocyte-derived nitric oxide in MS33. Finally, a classical gain of beneficial function is the restriction of immune cell infiltration in open injuries by scar-border forming reactive astrocytes7.
Transcriptomics and A1–A2 classification
Transcriptomics has contributed to a fundamental discovery: astrocytes in the healthy brain are diverse and specialized to perform specific roles in distinct CNS circuits14,65. Astrocyte diversity in healthy tissue arises from embryonic patterning programs or local neuronal cues14. Likewise, reactive astrocytes are also diverse, as unequivocally demonstrated by microarray-based66,67,68 and RNAseq-based48,69,70,71 transcriptomic profiling of mouse bulk astrocytes48,66,67,68,69,70 or of astrocyte populations preselected according to cell-surface markers71. Such transcriptomic profiling specifically shows that reactive astrocytes adopt distinct molecular states in different disease models48,66,67,68,69,70, in different CNS regions70, and in brain tumors71. These studies also suggested complex functional changes in reactive astrocytes, including novel regenerative functions70, proliferation, and neural stem cell potential68, as well as loss of homeostatic functions66. They have also identified drug candidates to establish the impact of altered astrocytic pathways in mouse models68,70. Whether baseline astrocyte heterogeneity influences astrocyte reactivity is an outstanding question.
In one early transcriptome study66 and its follow-up72, it was proposed that mouse astrocytes adopted an ‘A1’ neurotoxic phenotype after exposure to specific cytokines secreted by microglia exposed to lipopolysaccharide (LPS), whereas they acquire an ‘A2’ neuroprotective phenotype after ischemic stroke―two acute pathological conditions. Two correlative signatures of 12 genes with 14 pan-reactive genes were proposed as fingerprints identifying these phenotypes and, for A1 astrocytes, combined with thorough functional analyses in vitro72. Although the A1 and A2 phenotypes were not proposed to be universal or all-encompassing, they became widely misinterpreted as evidence for a binary polarization of reactive astrocytes in either neurotoxic or neuroprotective states, which could be readily identified in any CNS disease, acute or chronic, by their correlative marker genes in a manner similar to the once-popular, but now discarded, Th1–Th2 lymphocyte and M1–M2 microglia polarization theories73. For multiple reasons, we now collectively recommend moving beyond the A1–A2 labels and the misuse of their marker genes. Importantly, only a subset, often a mix of A1 and A2 or pan-reactive transcripts, are upregulated in astrocytes from brains of study participants with HD74 or AD75,76, or from several mouse models of acute injuries and chronic diseases of the CNS42,69,76,77. Moreover, the functions of these genes are not known, because, to date, no experimental evidence has causally linked any of the proposed marker genes of A1 or A2 astrocytes to either toxic or protective functions. Thus, the mere expression of some or even all these marker genes does not prove the presence of functions that these genes have not been demonstrated to exert. Specifically, complement factor 3 (C3) should not be regarded as a single and definitive marker that unequivocally labels astrocytes with a net detrimental effect. In addition, steadily increasing evidence indicates that any binary polarization of reactive astrocytes falls short of capturing their phenotypic diversity across disorders. For example, single-cell and single-nucleus RNAseq (scRNAseq and snRNAseq, respectively) studies in mouse models and human brains of chronic neurodegenerative diseases have unraveled numerous stage-dependent transcriptomic states in HD74, AD75,78, and MS40 that do not clearly comply with A1–A2 profiles. In addition, advanced statistics using multidimensional data and co-clustering approaches reveals that the A1 and A2 transcriptomes represent only two of many potential astrocyte transcriptomes segregating along several latent variables79. The analyses also indicate that multidimensional data are necessary to establish the distinctiveness of astrocyte phenotypes (Fig. 2). Characterization of the potentially extensive and subtle functional diversity of reactive astrocytes suggested by transcriptomic data is an important future goal.
Fig. 2: Workflow for the identification of key variables shaping astrocyte reactivity using multidimensional analyses.
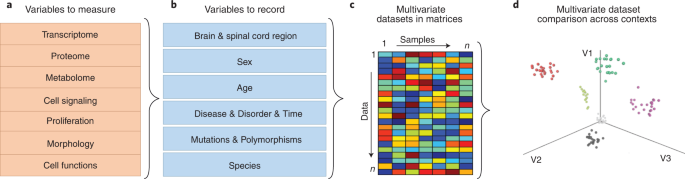
a, Variables to measure in individual experiments. Although at present it is unrealistic to measure all in the same experiment, it will in most cases be possible to measure at least two or three. b, Variables to record in individual experiments. In some experiments, all or most of these variables are kept constant and are not compared, but they should all be recorded to allow for future comparison across experiments and studies. c, Individual studies will generate multidimensional datasets of reactive astrocytes that can be organized in matrices containing all outcome measures of variables assessed in a (for example, -omics data, functional measurements). One matrix may be generated for each condition listed in b using data obtained in a. Determining whether such states are equivalent to fixed categories rather than temporary changes due to the dynamic nature of cell functioning requires cross-comparison among studies or longitudinal studies, paired with statistical analyses, as in d. d, Multidimensional data analysis and clustering statistics of weighted scores from datasets (a) across different contexts (b) represented in matrices (c) allow identification of functional vectors (V) driving astrocyte reactivity in different contexts. A high score and a low score in each vector represent gain and loss of function, respectively. The graph shows a hypothetical plot of simulated multivariate datasets from a (each dot represents one dataset or sample) obtained in different contexts (b), depicted in different colors. Astrocytes with shared features segregate together along three axes according to the predominance of the function represented in each vector. A state is defined by where the dataset(s) are placed in the V1-3 space. The analysis can be _n_-dimensional, but for visual clarity, we show a 3D scenario.
Human stem cells
Advances in human induced pluripotent stem cell (hiPSC) technology are being adapted to astrocyte research. Interestingly, astrocytes generated from hiPSC derived from fibroblasts obtained from patients with CNS diseases (usually with a genetic mutation causative of disease or a risk polymorphism) show pathological phenotypes, including dysregulation of lipid metabolism11, alteration in the contents of the extracellular vesicles released by astrocytes80, reduced autophagy81, or altered STAT3 signaling82. hiPSC-derived astrocytes are also amenable to study responses to viral infection83 and to specific stimuli84. Nevertheless, caution is in order, for more research is needed to establish hiPSC-derived astrocytes as bona fide models of human astrocytes and to determine whether they recapitulate the maturity as well as the temporal, regional, and individual heterogeneity of in vivo astrocytes. Importantly, not only are these cells removed from their original milieu, but the serum pervasively used in culture media may render them reactive84. In addition, generation of astrocytes from neural stem cells is inherently difficult, and derivation and culture conditions have not yet been standardized, leading to diversity of clone phenotypes. Finally, aging-related neurodegenerative diseases should be modeled with astrocytes derived from cells from aged individuals, but, in this case, the epigenetic rejuvenation intrinsic to the reprogramming of adult cells arises as a confounding factor to be controlled for.
Are aging astrocytes reactive or senescent?
Healthy brain aging is not pathological and may be defined as an adaptive evolution of global cell physiology over time85. Aged human brains display only mild and heterogeneous changes in astrocyte morphology or GFAP levels86. Studies in rodents document region-dependent and often contradictory changes in aging astrocytes, such as an increase in cellular volume and overlap of astrocyte processes, but also atrophy, increase in GFAP content, or even a reduction in the number of GFAP+ and GS+ astrocytes87,88,89. Notably, aging is also associated with pronounced regional differences in astrocyte gene expression in mouse brains90,91. However, only a few studies have directly assessed astrocyte functions in the aging mouse brain85,92. Thus, although the data suggest complex changes in aging astrocytes, the evidence is not yet sufficient to qualify astrocytes as being bona fide reactive during physiological aging. Nonetheless, with advanced age, cumulative exposure to pathological stimuli may render some astrocytes reactive. To test this hypothesis, a systematic investigation of the molecular properties of aging astrocytes across different CNS regions in humans and comparison of physiologically aged and reactive astrocytes in various pathological conditions are needed, together with functional validations in mouse models. Finally, we suggest caution about extending the concept of senescence to astrocytes based upon the expression of cell senescence marker p16INK4A, increased β-galactosidase activity, and secretion of cytokines93, because the core definition of senescence (that is, irreversible cell-cycle arrest in proliferative cells) may not apply to astrocytes, which are essentially post-mitotic cells that rarely divide in healthy tissue. Molecular and functional profiling of putative senescent astrocytes in different diseases is needed to clarify the meaning of p16INK4A expression in post-mitotic astrocytes, as well as the interplay between senescence-like features, reactivity, and aging in astrocytes.
Are new names needed?
Arguably, new names are needed to capture the variety of reactive astrocytes, but current knowledge does not yet allow the objective categorizing of reactive astrocytes. Indeed, the existence of fixed categories defined by molecular and functional features consistently observed in different disease contexts is not yet certain. Nonetheless, two new names have recently been coined to describe the extremes of six astrocytic transcriptional clusters detected by snRNAseq in the hippocampus of AD transgenic and wild-type mice78. In this study, ‘homeostatic astrocytes’ were predominant in healthy mice, whereas ‘disease-associated astrocytes’ were unique to AD mice. We do not support generalization of this ‘disease-associated’ classification to other conditions because only one disease was studied. In addition, the term ‘homeostatic astrocytes’ implies the unproven assumption that other transcriptional astrocyte clusters are dys-homeostatic, while they may be successful homeostasis-preserving adaptations to disease.
We stress that the expression in full or in part of a predetermined correlative signature of molecular markers is not, on its own, sufficient to define a functional phenotype of reactive astrocyte. In addition, vague and binary terms such as ‘neuroprotective’ or ‘neurotoxic’ are best avoided in describing astrocyte phenotypes as they are too simplistic to be meaningful, unless they are supported by specific molecular mechanisms and direct causative experimental evidence. Future classification of reactive astrocytes should, instead, consider multiple criteria including transcriptome, proteome, morphology, and specific cellular functions (Table 2), together with demonstrated impact on pathological hallmarks (Fig. 2).
For now, we recommend ‘reactive astrocytes’ as the general term for astrocytes observed in pathological conditions (Box 1). The term ‘injured’ or ‘wounded’ astrocytes should be reserved for astrocytes with unequivocal morphological signs of damage (for example, beaded processes), as observed in ischemia and trauma30,31. Descriptions based on misleading generalizations of functional changes and over-interpretation of correlative data should be avoided. We call for a clear operational terminology that includes information about morphology (for example, hypertrophic, atrophic), molecular markers (Table 1), and functional readouts (Table 2), as well as brain region, disease, disease stage, sex, species, and any other relevant source of heterogeneity (Fig. 2). Indeed, the goal is to go beyond the mere categorization of reactive astrocytes and identify the key variables driving specific reactive astrocyte states, phenotypes, and functions in specific contexts. When addressing similar issues for neurons, scientists are not concerned about categorizing disease-associated neurons into simple generalizable subtypes; rather, the emphasis is placed on understanding specific changes of defined neuronal populations in specific diseases. This principle should also apply to astrocytes.
Toward astrocyte-targeting therapies
One goal of research on reactive astrocytes is to develop astrocyte-targeting therapies for CNS diseases. Two challenges preclude translating the wealth of functional and molecular data described in the previous sections into therapies. First, there is a need to unequivocally clarify whether or not reactive astrocytes and their associated signaling pathways significantly contribute to the pathogenesis of specific CNS diseases. The approach should be reciprocal, such that human data inform experimental manipulations in animal models and animal data are validated in human materials. The second challenge is to develop astrocyte therapies tailored to specific disease contexts. Specific research directions include heterogeneity characterization, signaling, humanizing research, and systems biology.
Heterogeneity characterization
To define astrocyte phenotypes, all sources of heterogeneity should be considered and integrated with multidimensional statistical analyses (Fig. 2). ScRNAseq and snRNAseq are becoming established as valuable tools to gain insight into basal94 and reactive-astrocyte heterogeneity (Fig. 1e)40,78,95. Notably, isolation protocols may not always be optimal for astrocytes, resulting in low numbers of cells or nuclei being sequenced, and some highly relevant but weakly expressed transcripts, such as transcription factors and plasma-membrane receptors, may be overlooked, particularly in snRNAseq. Translation from scRNAseq or snRNAseq data to in situ immunohistochemical detection and functional validations is far from trivial, because the molecular profiles of astrocyte clusters and subpopulations partly overlap. Thus, instead of individual markers, signatures composed of a combination of markers with specified levels of expression or relative fold-changes are required to identify astrocyte phenotypes74. Such signatures must be statistically validated to the point of predicting phenotypes. Alternatively, the diversity within astrocyte populations from mouse models may be dissected out by combining fluorescence-activated cell sorting (FACS) and cell-surface markers identified in screens71. Further, emerging spatial transcriptomics that allow the simultaneous in situ detection of numerous genes will be of value to study the heterogeneity of reactive astrocytes at local and topographical levels (Fig. 1f)96. Importantly, molecular signatures based on the expression of genes or proteins need to be validated by assessing specific astrocyte functions (Table 2), since post-transcriptional and post-translational events critically shape functional outcomes. Functional validations should preferably be performed in vivo or using in vitro models closely mimicking human diseases. Classical knockout-, knockdown-, or CRISPR-based approaches to inactivate gene expression are available to gain insight into the impact on disease of a given pathway within previously identified astrocyte subsets40.
Signaling
An important implication of the disease-specific induction of distinct reactive astrocyte states is that the damage- and pathogen-associated stimuli from one disorder cannot be assumed to be active in another. For example, the now widely-used cocktail of factors released by LPS-treated neonatal microglia72 cannot be simply assumed to model reactive astrocytes in diseases other than neonatal septic shock due to infection by gram-negative bacteria. Likewise, exposure to Tau, amyloid-β, or α-synuclein needs to be carefully designed in vivo and in vitro to replicate the concentration, protein species, and combinations thereof found in patient brains. Acute metabolic damage with the mitochondrial toxin MPTP does not replicate chronic Parkinson’s disease (PD), to cite another example of in vivo inappropriate modelling. To complicate things further, the outcome of activating a signaling pathway may depend on the upstream stimuli82 or priming caused by previous exposure to other stimuli97, perhaps through epigenetic control40. Thus, careful selection of upstream stimuli is essential for appropriate in vivo and in vitro modelling of disease-specific reactive astrocytes. Finally, interventional strategies such as classical pharmacology56,98, genetic manipulation42,56, and biomaterials99 are available tools to modify pathological signaling in reactive astrocytes for therapeutic purposes. Optogenetics25 and designer receptors exclusively activated by designer drugs (DREADDs)25 are potential tools to manipulate reactive astrocytes or to restore their aberrant Ca2+ signaling observed in mouse models of neurodegenerative diseases53,54,55. However, it is unknown whether and how the changes in Na+, K+, Cl–, and Ca2+ fluxes and second messengers triggered by these approaches25 modulate signaling cascades driving phenotypical changes of reactive astrocytes (for example, JAK–STAT and NF-κB pathways)6.
Humanizing research
Although some basic functional properties of astrocytes have been shown to be evolutionarily conserved between humans and rodents100, it is still critical to study patient samples and develop models of human reactive astrocytes, because morphological and transcriptomic comparisons have revealed prominent differences between mice and humans101,102,103. In addition to astrocytes from postmortem samples and biopsies59 (Fig. 1b), hiPSC-derived astrocytes, which can be generated with a fast protocol in 2D layers104 or integrated in 3D systems such as spheroids and organoids105,106,107,108, are rapidly becoming commonplace in basic research11,82 and therapy development109. Researchers need to be aware of the pros and cons of the various protocols available, as discussed in previous sections and elsewhere110,111,112. Also, hiPSC glial mouse chimeric brains, in which hiPSC differentiate into human astrocytes, oligodendrocytes, and their progenitors, offer the possibility to study human astrocytes from patients in contexts amenable to in vivo experimentation113,114. In addition, proteins released by injured astrocytes are currently being considered as fluid biomarkers of neurological trauma31. Biomarkers of reactive astrocytes in human disease will indeed be needed to demonstrate target engagement of future astrocyte-directed therapies in clinical trials. Emerging reactive-astrocyte biomarkers are either measured in blood or cerebrospinal fluid (for example, YKL-40)115 or used for brain imaging, such as MAO-B-based positron emission tomography (PET)116, which provides important topographical information (Table 1)117. Plausibly, disease-specific biomarker signatures rather than single ubiquitous biomarkers will be needed.
Use of systems biology
Computerized tools, including systems biology and artificial intelligence, are essential to organizing and interpreting the increasing wealth of high-throughput, multidimensional molecular and functional data from reactive astrocytes. Currently, molecular data (for example, -omics) can be transformed into mathematical maps by artificial intelligence118, thereby providing quantitative representations of the otherwise-vague notion of phenotypes. An example of functional data is 2D and 3D Ca2+ imaging that generates kinetic profiles and maps for single astrocytes and 2D or 3D networks (Fig. 1c)119,120. Artificial intelligence can identify patterns of Ca2+ signaling in astrocytes55,120. Multidimensional molecular and functional data have then two applications. First, multivariate analysis may unravel molecules, pathways, and variables shaping astrocyte phenotypes in acute versus chronic degenerative conditions, different disease stages, sexes, and CNS regions (Fig. 2). Second, these data can be used to predict the net functional outcome of a complex mix of potentially protective or deleterious pathways and identification of hubs, such as master transcription factors or epigenetic regulators, that, when activated, promote globally beneficial transformations. Importantly, the inhibition of detrimental pathways must not secondarily impair protective ones or damage basic astrocyte functions. Finally, no astrocyte-targeting therapy can be successful if it does not consider the complex interactions of reactive astrocytes with other CNS cells.
Concluding remarks
The dawn of neuropathology in the late 19th and early 20th centuries witnessed widespread interest in neuroglia. Today, research on astrocytes and their remodeling in the context of injury, disease, and infection is undergoing a renaissance, with new researchers bringing exciting new techniques, approaches, and hypotheses. Given the scarcity of disease-modifying treatments for chronic diseases and acute injuries of the CNS, this astrocyte revival represents an opportunity to develop largely unexplored therapeutic niches, such as the manipulation of reactive astrocytes. However, despite the substantial body of knowledge accumulated since the discovery of reactive astrocytes a century ago, there are no therapies purposely designed against astrocyte-specific targets in clinical practice. The present working consensus for research guidelines will hopefully boost more coordinated and better focused efforts to improve and therapeutically exploit our knowledge about the role(s) of reactive astrocytes in CNS diseases and injuries.
References
- Virchow, R. Cellular Pathology (Robert M. De Witt, 1860).
- Achucarro, N. Some pathological findings in the neuroglia and in the ganglion cells of the cortex in senile conditions. Bull. Gov. Hosp. Insane 2, 81–90 (1910).
Google Scholar - Andriezen, W. L. The neuroglia elements in the human brain. Brit. Med. J. 2, 227–230 (1893). The first account of hypertrophic reactive astrocytes in pathology, although they were not called hypertrophic or reactive astrocytes.
Article CAS PubMed PubMed Central Google Scholar - Weigert, C. Beiträge zur Kenntnis der normalen menschlichen Neuroglia. in Zeitschrift für Psychologie und Physiologie der Sinnesorgane (Moritz Diesterweg, 1895).
- Del Río-Hortega, P. & Penfield, W. G. Cerebral cicatrix: The reaction of neuroglia and microglia to brain wounds. Bull. Johns Hopkins Hosp. 41, 278–303 (1927).
Google Scholar - Escartin, C., Guillemaud, O. & Carrillo-de Sauvage, M. A. Questions and (some) answers on reactive astrocytes. Glia 67, 2221–2247 (2019).
Article PubMed Google Scholar - Sofroniew, M. V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263 (2015).
Article CAS PubMed PubMed Central Google Scholar - Verkhratsky, A., Zorec, R. & Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol 27, 629–644 (2017).
Article CAS PubMed PubMed Central Google Scholar - Messing, A., Brenner, M., Feany, M. B., Nedergaard, M. & Goldman, J. E. Alexander disease. J. Neurosci. 32, 5017–5023 (2012).
Article CAS PubMed PubMed Central Google Scholar - Brusilow, S. W., Koehler, R. C., Traystman, R. J. & Cooper, A. J. Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 7, 452–470 (2010).
Article CAS PubMed PubMed Central Google Scholar - Lin, Y. T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7 (2018). Technically improved generation of hiPSC-derived astrocytes demonstrates that astrocytes harboring a genetic risk factor for AD are diseased astrocytes that may further exacerbate ongoing pathology.
Article CAS PubMed PubMed Central Google Scholar - Eng, L. F., Vanderhaeghen, J. J., Bignami, A. & Gerstl, B. An acidic protein isolated from fibrous astrocytes. Brain Res. 28, 351–354 (1971). The first identification of human GFAP in astrocytes from old multiple sclerosis plaques, post-leucotomy scars, and the occipital and frontal horns of the lateral ventricles in aged individuals with hydrocephalus ex vacuo.
Article CAS PubMed Google Scholar - Griemsmann, S. et al. Characterization of panglial gap junction networks in the thalamus, neocortex, and hippocampus reveals a unique population of glial cells. Cereb. Cortex 25, 3420–3433 (2015).
Article PubMed Google Scholar - Ben Haim, L. & Rowitch, D. H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 18, 31–41 (2017).
Article CAS PubMed Google Scholar - Kriegstein, A. & Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 (2009).
Article CAS PubMed PubMed Central Google Scholar - Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 (2008). This study represented a technical and conceptual breakthrough in the neurosciences as the first unbiased classification of brain cell populations based on transcriptomic profiles using early microarray analyses. The resulting transcriptomes are a powerful tool to gain insight into novel brain cell functions. More recently, the classification of brain cells has been further refined and enriched by sc/snRNAseq and spatial transcriptomics.
Article CAS PubMed PubMed Central Google Scholar - Roybon, L. et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep 4, 1035–1048 (2013).
Article CAS PubMed PubMed Central Google Scholar - Rossi, D. et al. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 15, 1691–1700 (2008).
Article CAS PubMed Google Scholar - Rodríguez, J. J., Terzieva, S., Olabarria, M., Lanza, R. G. & Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis 4, e678 (2013).
Article PubMed PubMed Central CAS Google Scholar - O’Callaghan, J. P., Brinton, R. E. & McEwen, B. S. Glucocorticoids regulate the synthesis of glial fibrillary acidic protein in intact and adrenalectomized rats but do not affect its expression following brain injury. J. Neurochem. 57, 860–869 (1991).
Article PubMed Google Scholar - Gerics, B., Szalay, F. & Hajós, F. Glial fibrillary acidic protein immunoreactivity in the rat suprachiasmatic nucleus: circadian changes and their seasonal dependence. J. Anat. 209, 231–237 (2006). Early demonstration that GFAP is regulated in a physiological context.
Article PubMed PubMed Central Google Scholar - Serrano-Pozo, A., Gómez-Isla, T., Growdon, J. H., Frosch, M. P. & Hyman, B. T. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol. 182, 2332–2344 (2013).
Article CAS PubMed PubMed Central Google Scholar - Wilhelmsson, U. et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc. Natl Acad. Sci. USA 103, 17513–17518 (2006). The complete visualization of astrocytes using whole-cell filling techniques revealed that reactive astrocytes display subtle morphological changes and remain in their 3D territorial domain, highlighting that GFAP immunostaining overestimates the true degree of astrocyte hypertrophy.
Article CAS PubMed PubMed Central Google Scholar - Sosunov, A. A. et al. Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J. Neurosci. 34, 2285–2298 (2014).
Article CAS PubMed PubMed Central Google Scholar - Yu, X., Nagai, J. & Khakh, B. S. Improved tools to study astrocytes. Nat. Rev. Neurosci. 21, 121–138 (2020).
Article CAS PubMed Google Scholar - Schiweck, J., Eickholt, B. J. & Murk, K. Important shapeshifter: mechanisms allowing astrocytes to respond to the changing nervous system during development, injury and disease. Front. Cell. Neurosci. 12, 261 (2018).
Article PubMed PubMed Central CAS Google Scholar - Olabarria, M., Noristani, H. N., Verkhratsky, A. & Rodríguez, J. J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838 (2010).
PubMed Google Scholar - Black, J. A., Newcombe, J. & Waxman, S. G. Astrocytes within multiple sclerosis lesions upregulate sodium channel Nav1.5. Brain 133, 835–846 (2010).
Article PubMed Google Scholar - Tachibana, M. et al. Clasmatodendrosis is associated with dendritic spines and does not represent autophagic astrocyte death in influenza-associated encephalopathy. Brain Dev 41, 85–95 (2019).
Article PubMed Google Scholar - Levine, J. et al. Traumatically injured astrocytes release a proteomic signature modulated by STAT3-dependent cell survival. Glia 64, 668–694 (2016).
Article PubMed Google Scholar - Halford, J. et al. New astroglial injury-defined biomarkers for neurotrauma assessment. J. Cereb. Blood Flow Metab. 37, 3278–3299 (2017). These data led to the first clinically used kit based on astrocyte-derived fluid biomarkers for neurotrauma assessments.
Article CAS PubMed PubMed Central Google Scholar - Ramon y Cajal, S. Contribución al conocimiento de la neuroglía del cerebro humano. Trabajos del Laboratorio de Investigaciones Biológicas de la Universidad de Madrid 11, 255–315 (1913).
Google Scholar - Colombo, E. et al. Stimulation of the neurotrophin receptor TrkB on astrocytes drives nitric oxide production and neurodegeneration. J. Exp. Med. 209, 521–535 (2012). Demonstration that astrocytes may become neurotoxic by releasing nitric oxide.
Article CAS PubMed PubMed Central Google Scholar - Theis, M. et al. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 23, 766–776 (2003).
Article CAS PubMed PubMed Central Google Scholar - Kraft, A. W. et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198 (2013).
Article CAS PubMed PubMed Central Google Scholar - Mucke, L. et al. Astroglial expression of human alpha(1)-antichymotrypsin enhances alzheimer-like pathology in amyloid protein precursor transgenic mice. Am. J. Pathol. 157, 2003–2010 (2000). Early demonstration in a mouse model of AD that targeted manipulation of astrocyte functions by transgenic tools has an impact on disease. A wealth of studies using transgenic mice and viral vectors followed suit and unequivocally demonstrate that reactive astrocytes influence CNS pathologies.
Article CAS PubMed PubMed Central Google Scholar - Xu, L., Emery, J. F., Ouyang, Y. B., Voloboueva, L. A. & Giffard, R. G. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia 58, 1042–1049 (2010).
Article PubMed PubMed Central Google Scholar - Furman, J. L. et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 16129–16140 (2012).
Article CAS PubMed PubMed Central Google Scholar - Pardo, L. et al. Targeted activation of CREB in reactive astrocytes is neuroprotective in focal acute cortical injury. Glia 64, 853–874 (2016).
Article PubMed Google Scholar - Wheeler, M. A. et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020). The first study combining scRNAseq to characterize reactive astrocytes with targeted molecular manipulations demonstrates, in a mouse model of MS, that reactive astrocytes are molecularly and functionally heterogeneous, depending on brain area and disease stage.
Article CAS PubMed PubMed Central Google Scholar - Bush, T. G. et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23, 297–308 (1999). The first demonstration that ablation of proliferative reactive astrocytes after stab wound injury in the mouse forebrain is deleterious. This study made the case that astrocyte reactivity is not always detrimental as widely believed, but may, instead, serve important homeostatic functions.
Article CAS PubMed Google Scholar - Ceyzériat, K. et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 6, 104 (2018).
Article PubMed PubMed Central CAS Google Scholar - Reichenbach, N. et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 11, e9665 (2019).
Article PubMed PubMed Central CAS Google Scholar - Kamphuis, W. et al. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63, 1036–1056 (2015).
Article PubMed Google Scholar - Wheeler, M. A. & Quintana, F. J. Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb. Perspect. Med. 9, a029009 (2019).
Article CAS PubMed PubMed Central Google Scholar - Colombo, E. & Farina, C. Astrocytes: key regulators of neuroinflammation. Trends Immunol 37, 608–620 (2016).
Article CAS PubMed Google Scholar - Nobuta, H. et al. STAT3-mediated astrogliosis protects myelin development in neonatal brain injury. Ann. Neurol. 72, 750–765 (2012).
Article CAS PubMed PubMed Central Google Scholar - Anderson, M. A. et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016).
Article CAS PubMed PubMed Central Google Scholar - Herrmann, J. E. et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 28, 7231–7243 (2008).
Article CAS PubMed PubMed Central Google Scholar - Tyzack, G. E. et al. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun. 5, 4294 (2014).
Article CAS PubMed Google Scholar - Santello, M., Toni, N. & Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 22, 154–166 (2019).
Article CAS PubMed Google Scholar - Semyanov, A., Henneberger, C. & Agarwal, A. Making sense of astrocytic calcium signals - from acquisition to interpretation. Nat. Rev. Neurosci. 21, 551–564 (2020).
Article CAS PubMed Google Scholar - Jiang, R., Diaz-Castro, B., Looger, L. L. & Khakh, B. S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 36, 3453–3470 (2016).
Article CAS PubMed PubMed Central Google Scholar - Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T. & Bacskai, B. J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215 (2009).
Article CAS PubMed PubMed Central Google Scholar - Agarwal, A. et al. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron 93, 587–605.e7 (2017). Technically refined application of Ca 2+ imaging approaches and machine learning unraveled dysregulation of Ca 2+ responses in a mouse model of ALS.
- Reichenbach, N. et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 215, 1649–1663 (2018).
Article CAS PubMed PubMed Central Google Scholar - Habbas, S. et al. Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell 163, 1730–1741 (2015). This study illustrates how modulation of astrocyte signaling via TNFα can switch from physiological to pathological.
Article CAS PubMed Google Scholar - Tong, X. et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703 (2014). Demonstration with targeted molecular manipulations that loss of astrocyte homeostatic functions contributes to HD pathogenesis.
Article CAS PubMed PubMed Central Google Scholar - Bedner, P. et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222 (2015).
Article PubMed PubMed Central Google Scholar - Gomez-Arboledas, A. et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66, 637–653 (2018).
Article PubMed Google Scholar - Le Douce, J. et al. Impairment of glycolysis-derived L-serine production in astrocytes contributes to cognitive deficits in Alzheimer’s disease. Cell Metab 31, 503–517.e8 (2020).
Article PubMed CAS Google Scholar - Zhang, M. et al. Lactate deficit in an Alzheimer disease mouse model: the relationship with neuronal damage. J. Neuropathol. Exp. Neurol. 77, 1163–1176 (2018).
Article CAS PubMed Google Scholar - Jo, S. et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896 (2014). Demonstration of astrocyte-targeted pharmacological manipulations to restore neural circuit homeostasis by correcting production of GABA by astrocytes in an AD mouse model.
Article CAS PubMed PubMed Central Google Scholar - Heo, J. Y. et al. Aberrant tonic inhibition of dopaminergic neuronal activity causes motor symptoms in animal models of Parkinson’s disease. Curr. Biol. 30, 276–291.e9 (2020).
Article CAS PubMed Google Scholar - Chai, H. et al. Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531–549.e9 (2017).
Article CAS PubMed PubMed Central Google Scholar - Zamanian, J. L. et al. Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 (2012). First evidence for molecular heterogeneity of reactive astrocytes using microarray-based transcriptomics of acutely isolated astrocytes from mouse models of ischemia and septic shock. Studies in virtually all models of CNS diseases followed.
Article CAS PubMed PubMed Central Google Scholar - Orre, M. et al. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 35, 2746–2760 (2014).
Article CAS PubMed Google Scholar - Sirko, S. et al. Astrocyte reactivity after brain injury-: The role of galectins 1 and 3. Glia 63, 2340–2361 (2015).
Article PubMed PubMed Central Google Scholar - Diaz-Castro, B., Gangwani, M. R., Yu, X., Coppola, G. & Khakh, B. S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 11, eaaw8546 (2019).
Article CAS PubMed Google Scholar - Itoh, N. et al. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl Acad. Sci. USA 115, E302–E309 (2018).
Article CAS PubMed Google Scholar - John Lin, C. C. et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405 (2017).
Article CAS PubMed Google Scholar - Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Article CAS PubMed PubMed Central Google Scholar - Ransohoff, R. M. A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991 (2016).
Article CAS PubMed Google Scholar - Al-Dalahmah, O. et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 8, 19 (2020).
Article CAS PubMed PubMed Central Google Scholar - Grubman, A. et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097 (2019).
Article CAS PubMed Google Scholar - Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Article CAS PubMed PubMed Central Google Scholar - Das, S., Li, Z., Noori, A., Hyman, B. T. & Serrano-Pozo, A. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflammation 17, 227 (2020).
Article CAS PubMed PubMed Central Google Scholar - Habib, N. et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020).
Article CAS PubMed Google Scholar - Henrik Heiland, D. et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 10, 2541 (2019).
Article PubMed PubMed Central CAS Google Scholar - Varcianna, A. et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635 (2019).
Article PubMed PubMed Central Google Scholar - di Domenico, A. et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Reports 12, 213–229 (2019).
Article PubMed PubMed Central CAS Google Scholar - Tyzack, G. E. et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat. Commun. 8, 1164 (2017).
Article PubMed PubMed Central CAS Google Scholar - Ledur, P. F. et al. Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci. Rep. 10, 1218 (2020).
Article CAS PubMed PubMed Central Google Scholar - Perriot, S. et al. Human induced pluripotent stem cell-derived astrocytes are differentially activated by multiple sclerosis-associated cytokines. Stem Cell Reports 11, 1199–1210 (2018).
Article CAS PubMed PubMed Central Google Scholar - Rodríguez-Arellano, J. J., Parpura, V., Zorec, R. & Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182 (2016).
Article PubMed CAS Google Scholar - Jyothi, H. J. et al. Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiol. Aging 36, 3321–3333 (2015).
Article CAS PubMed Google Scholar - Rodríguez, J. J. et al. Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23 (2014).
Article PubMed CAS Google Scholar - Cerbai, F. et al. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS One 7, e45250 (2012).
Article CAS PubMed PubMed Central Google Scholar - O’Callaghan, J. P. & Miller, D. B. The concentration of glial fibrillary acidic protein increases with age in the mouse and rat brain. Neurobiol. Aging 12, 171–174 (1991).
Article PubMed Google Scholar - Boisvert, M. M., Erikson, G. A., Shokhirev, M. N. & Allen, N. J. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22, 269–285 (2018).
Article CAS PubMed PubMed Central Google Scholar - Clarke, L. E. et al. Normal aging induces A1-like astrocyte reactivity. Proc. Natl Acad. Sci. USA 115, E1896–E1905 (2018).
CAS PubMed PubMed Central Google Scholar - Peters, O. et al. Astrocyte function is modified by Alzheimer’s disease-like pathology in aged mice. J. Alzheimers Dis. 18, 177–189 (2009).
Article CAS PubMed Google Scholar - Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017).
Article CAS PubMed PubMed Central Google Scholar - Batiuk, M. Y. et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11, 1220 (2020).
Article CAS PubMed PubMed Central Google Scholar - Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). First snRNAseq analysis in human AD samples identifies sub-populations of reactive astrocytes.
Article CAS PubMed PubMed Central Google Scholar - Chen, W. T. et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s Disease. Cell 182, 976–991.e19 (2020).
Article CAS PubMed Google Scholar - Hennessy, E., Griffin, E. W. & Cunningham, C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J. Neurosci. 35, 8411–8422 (2015).
Article CAS PubMed PubMed Central Google Scholar - Park, J. H. et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 5, v0316 (2019).
Article CAS Google Scholar - Zuidema, J. M., Gilbert, R. J. & Gottipati, M. K. Biomaterial approaches to modulate reactive astroglial response. Cells Tissues Organs 205, 372–395 (2018).
Article PubMed Google Scholar - Bedner, P., Jabs, R. & Steinhäuser, C. Properties of human astrocytes and NG2 glia. Glia 68, 756–767 (2020).
Article PubMed Google Scholar - Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016). First study reporting transcriptomes of human astrocytes, paving the way for the highly used open-source database of gene expression for all brain cell types in humans and mice (https://www.brainrnaseq.org/).
Article CAS PubMed Google Scholar - Oberheim, N. A. et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287 (2009).
Article CAS PubMed PubMed Central Google Scholar - Oberheim, N. A., Wang, X., Goldman, S. & Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 29, 547–553 (2006).
Article CAS PubMed Google Scholar - Tchieu, J. et al. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 37, 267–275 (2019).
Article CAS PubMed PubMed Central Google Scholar - Sloan, S. A. et al. Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron 95, 779–790.e6 (2017).
Article CAS PubMed PubMed Central Google Scholar - Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Article CAS PubMed Google Scholar - Quadrato, G. et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53 (2017).
Article CAS PubMed PubMed Central Google Scholar - Giandomenico, S. L. et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 22, 669–679 (2019).
Article CAS PubMed PubMed Central Google Scholar - Colombo, E. et al. Siponimod (BAF312) activates Nrf2 while hampering NFκB in human astrocytes, and protects from astrocyte-induced neurodegeneration. Front. Immunol. 11, 635 (2020).
Article CAS PubMed PubMed Central Google Scholar - Hirbec, H. et al. Emerging technologies to study glial cells. Glia 68, 1692–1728 (2020).
Article PubMed Google Scholar - Guttenplan, K. A. & Liddelow, S. A. Astrocytes and microglia: Models and tools. J. Exp. Med. 216, 71–83 (2019).
Article CAS PubMed PubMed Central Google Scholar - Almad, A. & Maragakis, N. J. A stocked toolbox for understanding the role of astrocytes in disease. Nat. Rev. Neurol. 14, 351–362 (2018).
Article PubMed Google Scholar - Han, X. et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353 (2013).
Article CAS PubMed PubMed Central Google Scholar - Osipovitch, M. et al. Human ESC-derived chimeric mouse models of Huntington’s disease reveal cell-Intrinsic defects in glial progenitor cell differentiation. Cell Stem Cell 24, 107–122.e7 (2019).
Article CAS PubMed Google Scholar - Craig-Schapiro, R. et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol. Psychiatry 68, 903–912 (2010).
Article CAS PubMed PubMed Central Google Scholar - Carter, S. F. et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 53, 37–46 (2012). Non-invasive imaging of reactive astrocytes in human patients.
Article CAS PubMed Google Scholar - Carter, S. F. et al. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med. 25, 77–95 (2019).
Article CAS PubMed Google Scholar - Romeo-Guitart, D. et al. Neuroprotective drug for nerve trauma revealed using artificial intelligence. Sci. Rep. 8, 1879 (2018).
Article PubMed PubMed Central CAS Google Scholar - Bindocci, E. et al. Three-dimensional Ca2+ imaging advances understanding of astrocyte biology. Science 356, eaai8185 (2017).
Article PubMed CAS Google Scholar - Wang, Y. et al. Accurate quantification of astrocyte and neurotransmitter fluorescence dynamics for single-cell and population-level physiology. Nat. Neurosci. 22, 1936–1944 (2019).
Article CAS PubMed PubMed Central Google Scholar - Ben Haim, L. et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 35, 2817–2829 (2015).
Article PubMed PubMed Central Google Scholar - Hol, E. M. & Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 32, 121–130 (2015).
Article CAS PubMed Google Scholar - Moreels, M., Vandenabeele, F., Dumont, D., Robben, J. & Lambrichts, I. Alpha-smooth muscle actin (alpha-SMA) and nestin expression in reactive astrocytes in multiple sclerosis lesions: potential regulatory role of transforming growth factor-beta 1 (TGF-beta1). Neuropathol. Appl. Neurobiol. 34, 532–546 (2008).
Article CAS PubMed Google Scholar - Jing, R. et al. Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J. Cell Sci. 120, 1267–1277 (2007).
Article CAS PubMed Google Scholar - Yamada, T., Kawamata, T., Walker, D. G. & McGeer, P. L. Vimentin immunoreactivity in normal and pathological human brain tissue. Acta Neuropathol. 84, 157–162 (1992).
Article CAS PubMed Google Scholar - Gui, Y., Marks, J. D., Das, S., Hyman, B. T. & Serrano-Pozo, A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol 30, 151–164 (2020).
Article CAS PubMed Google Scholar - Wilhelmus, M. M. et al. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 32, 119–130 (2006).
Article CAS PubMed Google Scholar - Furman, J. L. et al. Blockade of astrocytic calcineurin/NFAT signaling helps to normalize hippocampal synaptic function and plasticity in a rat model of traumatic brain injury. J. Neurosci. 36, 1502–1515 (2016).
Article CAS PubMed PubMed Central Google Scholar - Michetti, F. et al. The S100B story: from biomarker to active factor in neural injury. J. Neurochem. 148, 168–187 (2019).
Article CAS PubMed Google Scholar - Sun, W. et al. SOX9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J. Neurosci. 37, 4493–4507 (2017).
Article CAS PubMed PubMed Central Google Scholar - Wanner, I. B. et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886 (2013).
Article CAS PubMed PubMed Central Google Scholar - Campbell, S. C. et al. Potassium and glutamate transport is impaired in scar-forming tumor-associated astrocytes. Neurochem. Int. 133, 104628 (2020).
Article CAS PubMed Google Scholar - Voss, C. M. et al. AMP-activated protein kinase (AMPK) regulates astrocyte oxidative metabolism by balancing TCA cycle dynamics. Glia 68, 1824–1839 (2020).
Article PubMed Google Scholar - Kimbrough, I. F., Robel, S., Roberson, E. D. & Sontheimer, H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138, 3716–3733 (2015).
Article PubMed PubMed Central Google Scholar - Deshpande, T. et al. Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia 65, 1809–1820 (2017).
Article PubMed Google Scholar - Frakes, A. E. et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023 (2014).
Article CAS PubMed PubMed Central Google Scholar - Eraso-Pichot, A. et al. GSEA of mouse and human mitochondriomes reveals fatty acid oxidation in astrocytes. Glia 66, 1724–1735 (2018).
Article PubMed Google Scholar - Mächler, P. et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23, 94–102 (2016).
Article PubMed CAS Google Scholar - Lerchundi, R., Huang, N. & Rose, C. R. Quantitative imaging of changes in astrocytic and neuronal adenosine triphosphate using two different variants of ATeam. Front. Cell. Neurosci. 14, 80 (2020).
Article CAS PubMed PubMed Central Google Scholar - Ioannou, M. S. et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535.e14 (2019).
Article CAS PubMed Google Scholar - Polyzos, A. A. et al. Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab 29, 1258–1273.e11 (2019).
Article CAS PubMed PubMed Central Google Scholar - Oe, Y., Akther, S. & Hirase, H. Regional distribution of glycogen in the mouse brain visualized by immunohistochemistry. Adv. Neurobiol 23, 147–168 (2019).
Article PubMed Google Scholar - Vezzoli, E. et al. Ultrastructural evidence for a role of astrocytes and glycogen-derived lactate in learning-dependent synaptic stabilization. Cereb. Cortex 30, 2114–2127 (2020).
Article CAS PubMed Google Scholar - Vicente-Gutierrez, C. et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 1, 201–211 (2019).
Article CAS PubMed Google Scholar - Damisah, E. C. et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 6, a3239 (2020).
Article CAS Google Scholar - Simonovitch, S. et al. Impaired autophagy in APOE4 astrocytes. J. Alzheimers Dis. 51, 915–927 (2016).
Article CAS PubMed Google Scholar - Goetzl, E. J. et al. Traumatic brain injury increases plasma astrocyte-derived exosome levels of neurotoxic complement proteins. FASEB J. 34, 3359–3366 (2020).
Article CAS PubMed Google Scholar - Orre, M. et al. Reactive glia show increased immunoproteasome activity in Alzheimer’s disease. Brain 136, 1415–1431 (2013).
Article PubMed Google Scholar - Sirko, S. et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell 12, 426–439 (2013).
Article CAS PubMed Google Scholar - Buffo, A. et al. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl Acad. Sci. USA 105, 3581–3586 (2008).
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
Funding: CNRS, CEA, ANR, and France Alzheimer to CE.; MCINN (PID2019-107633RB-I00) and Generalitat de Catalunya (2017-SGR547, Grup de demències Sant Pau) to E.G. US Centers for Disease Control and Prevention to J. P.O. Alzheimer’s Association (AACF-17-524184) and NIH-NIA (K08AG064039) to A.S.-P. DFG (SPP1757, STE 552/5, STE 552/4), EU (H2020-MSCA-ITN project 722053 EU-GliaPhD) and BMBF (16GW0182 CONNEXIN) to C.S. Swiss National Science Foundation grant 31003A 173124/1; SNSF NCCR ‘Transcure’ (51NF40-160620); Synapsis Foundation Heidi Seiler-Stiftung 2018-PI01 to A.Volterra. NIH-NINDS (NS084030), Dr. Miriam and Sheldon G. Adelson Medical Foundation and Wings for Life to M.V.S. The authors thank T. Yohannan of Alpha Language Services, Barcelona, for expert copy editing.
Author information
Author notes
- These authors contributed equally: András Lakatos, James P. O’Callaghan, Gabor C. Petzold, Alberto Serrano-Pozo, Christian Steinhauser, Andrea Volterra, Giorgio Carmignoto.
- These authors jointly supervised this work: Carole Escartin, Elena Galea, Michael V. Sofroniew, Alexei Verkhratsky.
Authors and Affiliations
- Université Paris-Saclay, CEA, CNRS, MIRCen, Laboratoire des Maladies Neurodégénératives, Fontenay-aux-Roses, France
Carole Escartin, Gilles Bonvento & Miriam Riquelme-Perez - Institut de Neurociències and Departament de Bioquímica i Biologia Molecular, Unitat de Bioquímica de Medicina, Universitat Autònoma de Barcelona, Barcelona, Spain
Elena Galea - ICREA, Barcelona, Spain
Elena Galea - John van Geest Centre for Brain Repair and Division of Stem Cell Neurobiology, Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK
András Lakatos - Wellcome Trust-MRC Cambridge Stem Cell Institute, Cambridge Biomedical Campus, Cambridge, UK
András Lakatos & David H. Rowitch - Health Effects Laboratory Division, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, Morgantown, West Virginia, USA
James P. O’Callaghan - German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
Gabor C. Petzold - Division of Vascular Neurology, Department of Neurology, University Hospital Bonn, Bonn, Germany
Gabor C. Petzold - Alzheimer Research Unit, Department of Neurology, Massachusetts General Hospital, Charlestown, Massachusetts, USA
Alberto Serrano-Pozo - Harvard Medical School, Boston, Massachusetts, USA
Alberto Serrano-Pozo - Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, Bonn, Germany
Christian Steinhäuser - Department of Fundamental Neuroscience, University of Lausanne, Lausanne, Switzerland
Andrea Volterra - Neuroscience Institute, Italian National Research Council (CNR), Padua, Italy
Giorgio Carmignoto - Department of Biomedical Sciences, University of Padua, Padua, Italy
Giorgio Carmignoto - The Chica and Heinz Schaller Research Group, Institute for Anatomy and Cell Biology, Heidelberg University, Heidelberg, Germany
Amit Agarwal - Salk Institute for Biological Studies, Molecular Neurobiology Laboratory, La Jolla, California, USA
Nicola J. Allen - Department of Neuroscience, University of Minnesota, Minneapolis, Minnesota, USA
Alfonso Araque - Institut Pasteur de Montevideo, Montevideo, Uruguay
Luis Barbeito - Department of Neurobiology, George S. Wise, Faculty of Life Sciences and Sagol School of Neuroscience, Tel Aviv University, Ramat Aviv Tel Aviv, Israel
Ari Barzilai - The Solomon H. Snyder Department of Neuroscience, Kavli Neuroscience Discovery Institute, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
Dwight E. Bergles - School of Pharmacy and Biomedical Science, University of Portsmouth, Portsmouth, UK
Arthur M. Butt - Center for Brain and Disease Research, VIB and University of Leuven, Leuven, Belgium
Wei-Ting Chen & Bart De Strooper - ‘Physiology and Physiopathology of the Gliovascular Unit’ Research Group, Center for Interdisciplinary Research in Biology (CIRB), College de France, Unité Mixte de Recherche 7241 CNRS, Unité1050 INSERM, PSL Research University, Paris, France
Martine Cohen-Salmon - Trinity Biomedical Sciences Institute & Trinity College Institute of Neuroscience, School of Biochemistry & Immunology, Trinity College Dublin, Dublin, Republic of Ireland
Colm Cunningham - Center for Cell and Gene Therapy, Department of Neurosurgery, Baylor College of Medicine, Houston, Texas, USA
Benjamin Deneen - UK Dementia Research Institute at the University College London, London, UK
Bart De Strooper - UK Dementia Research Institute at the University of Edinburgh, Centre for Discovery Brain Sciences, Edinburgh, UK
Blanca Díaz-Castro - Institute of Experimental Neurology (INSpe) and Division of Neuroscience, San Raffaele Scientific Institute, Milan, Italy
Cinthia Farina - Vollum Institute, OHSU, Portland, Oregon, USA
Marc Freeman - Center for Neuroscience Research, Children’s National Research Institute, Children’s National Hospital, Washington DC, USA
Vittorio Gallo - Department of Pathology & Cell Biology, Columbia University, New York, New York, USA
James E. Goldman - University of Rochester Medical Center, Rochester, New York, USA
Steven A. Goldman - Center for Translational Neuromedicine, University of Copenhagen Faculty of Health and Medical Science and Rigshospitalet, Kobenhavn N, Denmark
Steven A. Goldman - Physiological Genomics, Biomedical Center, Ludwig-Maximilians-Universitaet & Institute of Stem Cell Research, Helmholtz Center Munich, Munich, Germany
Magdalena Götz - Synergy, Excellence Cluster of Systems Neurology, Biomedical Center, Munich, Germany
Magdalena Götz - Dpto. Biología Celular, Genética y Fisiología, Instituto de Investigación Biomédica de Málaga-IBIMA, Facultad de Ciencias, Universidad de Málaga, Málaga, Spain
Antonia Gutiérrez - Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), Madrid, Spain
Antonia Gutiérrez & Javier Vitorica - Department of Neuroscience, Tufts University School of Medicine, Boston, Massachusetts, USA
Philip G. Haydon - Microenvironment and Immunology Research Laboratory, Medical Center, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Dieter H. Heiland - Department of Neurosurgery, Medical Center, University of Freiburg, Faculty of Medicine, Freiburg, Germany
Dieter H. Heiland - Department of Translational Neuroscience, University Medical Center Utrecht Brain Center, Utrecht University, Utrecht, The Netherlands
Elly M. Hol - Laboratory of Glia Biology, VIB-KU Leuven Center for Brain and Disease Research, Leuven, Belgium
Matthew G. Holt - Division of Cellular and Molecular Pharmacology, Nihon University School of Medicine, Tokyo, Japan
Masamitsu Iino - Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts, USA
Ksenia V. Kastanenka - Cellular Neurosciences, Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
Helmut Kettenmann - Department of Physiology, David Geffen School of Medicine at UCLA, Los Angeles, California, USA
Baljit S. Khakh - Department of Neuropharmacology, Interdisciplinary Graduate School of Medicine, University of Yamanashi, Yamanashi, Japan
Schuichi Koizumi - Center for Cognition and Sociality, Institute for Basic Science 55, Expo-ro, Yuseong-gu, Daejeon, Korea
C. Justin Lee - Neuroscience Institute, Department of Neuroscience and Physiology, Department of Ophthalmology, NYU School of Medicine, New York, USA
Shane A. Liddelow - Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, British Columbia, Canada
Brian A. MacVicar - Division of Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
Pierre Magistretti - Centre de Neurosciences Psychiatriques, University of Lausanne and CHUV, Site de Cery, Prilly-Lausanne, Lausanne, Switzerland
Pierre Magistretti - Waisman Center and School of Veterinary Medicine, University of Wisconsin-Madison, Madison, Wisconsin, USA
Albee Messing - Department of Neurology Jungers Center for Neurosciences Research and Knight Cardiovascular Institute, Oregon Health & Science University, Portland, Oregon, USA
Anusha Mishra - Departments of Psychiatry/Weill Institute for Neuroscience University of California, San Francisco, California, USA
Anna V. Molofsky - Centre for Research in Neuroscience, Department of Neurology & Neurosurgery, Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada
Keith K. Murai - Sanders-Brown Center on Aging, University of Kentucky College of Medicine, Lexington, Kentucky, USA
Christopher M. Norris - Department of Immunobiology and Neuroscience, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan
Seiji Okada - Université de Bordeaux, Inserm, Neurocentre Magendie, U1215, Bordeaux, France
Stéphane H. R. Oliet & Aude Panatier - Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
João F. Oliveira - ICVS/3B’s -PT Government Associate Laboratory, Braga/Guimarães, Portugal
João F. Oliveira - IPCA-EST-2Ai, Polytechnic Institute of Cávado and Ave, Applied Artificial Intelligence Laboratory, Campus of IPCA, Barcelos, Portugal
João F. Oliveira - Department of Neurobiology, The University of Alabama at Birmingham, Birmingham, Alabama, USA
Vladimir Parpura - Laboratory of Regenerative Neuroimmunology, Center for Brain Repair, Department of Clinical Neuroscience, Institute of Neuroscience and Physiology, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden
Marcela Pekna - Laboratory of Astrocyte Biology and CNS Regeneration, Center for Brain Repair, Department of Clinical Neuroscience, Institute of Neuroscience and Physiology, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden
Milos Pekny - INSERM U1082, Université de Poitiers, Poitiers, France
Luc Pellerin - Department of Functional and Systems Neurobiology, Cajal Institute, CSIC, Madrid, Spain
Gertrudis Perea - Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology and Neuroscience, King’s College London, London, UK
Beatriz G. Pérez-Nievas - Centre National de la Recherche Scientifique, Université de Strasbourg, Institut des Neurosciences Cellulaires et Intégratives, Strasbourg, France
Frank W. Pfrieger - Department of Biochemistry & Biophysics, Kavli Institute for Fundamental Neuroscience, University of California, San Francisco, California, USA
Kira E. Poskanzer - Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School. Associate Member, The Broad Institute, Boston, Massachusetts, USA
Francisco J. Quintana - Third Rock Ventures, Boston, Massachusetts, USA
Richard M. Ransohoff - Fralin Biomedical Research Institute at Virginia Tech Carilion, School of Neuroscience Virginia Tech, Riverside Circle, Roanoke, Virginia, USA
Stefanie Robel - Institute of Neurobiology, Heinrich Heine University, Düsseldorf, Germany
Christine R. Rose - Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
Jeffrey D. Rothstein - Neuroglial Interactions in Cerebral Physiology and Pathologies, Center for Interdisciplinary Research in Biology, Collège de France, CNRS UMR 7241, INSERM U1050, Labex Memolife, PSL Research University Paris, Paris, France
Nathalie Rouach - Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow, Russia
Alexey Semyanov - Sechenov First Moscow State Medical University, Moscow, Russia
Alexey Semyanov - Physiological Genomics, Biomedical Center, LMU Munich, Munich, Germany
Swetlana Sirko - Institute for Stem Cell Research, Helmholtz Zentrum Munich, Neuherberg, Germany
Swetlana Sirko - Virginia Tech School of Neuroscience and Center for Glial Biology in Health, Disease and Cancer, Virginia Tech at the Fralin Biomedical Research Institute, Roanoke, Virginia, USA
Harald Sontheimer - Dept. of Neurology, University of California San Francisco and San Francisco Veterans Affairs Health Care System, San Francisco, California, USA
Raymond A. Swanson - Dept. Bioquímica y Biología Molecular, Instituto de Biomedicina de Sevilla, Universidad de Sevilla, Hospital Virgen del Rocío/CSIC, Sevilla, Spain
Javier Vitorica - Semel Institute for Neuroscience & Human Behavior, IDDRC, David Geffen School of Medicine, UCLA, Los Angeles, California, USA
Ina-Beate Wanner - George W. Woodruff School of Mechanical Engineering, Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory, and Parker H. Petit Institute for Bioengineering & Bioscience, Georgia Institute of Technology, Atlanta, Georgia, USA
Levi B. Wood - The Vivian L. Smith Department of Neurosurgery, Center for Stem Cell and Regenerative Medicine, MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, McGovern Medical School, UTHealth, University of Texas Health Science Center at Houston, Houston, TX, USA
Jiaqian Wu - Department of Neurosciences, UC San Diego School of Medicine, La Jolla; VA San Diego Research Service, San Diego, CA, USA
Binhai Zheng - Department of Pharmacology, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil
Eduardo R. Zimmer - Laboratory of Neuroendocrinology, Molecular Cell Physiology, Institute of Pathophysiology, University of Ljubljana, Faculty of Medicine, Ljubljana, Slovenia
Robert Zorec - Celica Biomedical, 1000, Ljubljana, Slovenia
Robert Zorec - Department of Neurobiology, David Geffen School of Medicine, University of California, Los Angeles, California, USA
Michael V. Sofroniew - Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, UK
Alexei Verkhratsky - Achúcarro Basque Center for Neuroscience, IKERBASQUE, Basque Foundation for Science, Bilbao, Spain
Alexei Verkhratsky
Authors
- Carole Escartin
You can also search for this author inPubMed Google Scholar - Elena Galea
You can also search for this author inPubMed Google Scholar - András Lakatos
You can also search for this author inPubMed Google Scholar - James P. O’Callaghan
You can also search for this author inPubMed Google Scholar - Gabor C. Petzold
You can also search for this author inPubMed Google Scholar - Alberto Serrano-Pozo
You can also search for this author inPubMed Google Scholar - Christian Steinhäuser
You can also search for this author inPubMed Google Scholar - Andrea Volterra
You can also search for this author inPubMed Google Scholar - Giorgio Carmignoto
You can also search for this author inPubMed Google Scholar - Amit Agarwal
You can also search for this author inPubMed Google Scholar - Nicola J. Allen
You can also search for this author inPubMed Google Scholar - Alfonso Araque
You can also search for this author inPubMed Google Scholar - Luis Barbeito
You can also search for this author inPubMed Google Scholar - Ari Barzilai
You can also search for this author inPubMed Google Scholar - Dwight E. Bergles
You can also search for this author inPubMed Google Scholar - Gilles Bonvento
You can also search for this author inPubMed Google Scholar - Arthur M. Butt
You can also search for this author inPubMed Google Scholar - Wei-Ting Chen
You can also search for this author inPubMed Google Scholar - Martine Cohen-Salmon
You can also search for this author inPubMed Google Scholar - Colm Cunningham
You can also search for this author inPubMed Google Scholar - Benjamin Deneen
You can also search for this author inPubMed Google Scholar - Bart De Strooper
You can also search for this author inPubMed Google Scholar - Blanca Díaz-Castro
You can also search for this author inPubMed Google Scholar - Cinthia Farina
You can also search for this author inPubMed Google Scholar - Marc Freeman
You can also search for this author inPubMed Google Scholar - Vittorio Gallo
You can also search for this author inPubMed Google Scholar - James E. Goldman
You can also search for this author inPubMed Google Scholar - Steven A. Goldman
You can also search for this author inPubMed Google Scholar - Magdalena Götz
You can also search for this author inPubMed Google Scholar - Antonia Gutiérrez
You can also search for this author inPubMed Google Scholar - Philip G. Haydon
You can also search for this author inPubMed Google Scholar - Dieter H. Heiland
You can also search for this author inPubMed Google Scholar - Elly M. Hol
You can also search for this author inPubMed Google Scholar - Matthew G. Holt
You can also search for this author inPubMed Google Scholar - Masamitsu Iino
You can also search for this author inPubMed Google Scholar - Ksenia V. Kastanenka
You can also search for this author inPubMed Google Scholar - Helmut Kettenmann
You can also search for this author inPubMed Google Scholar - Baljit S. Khakh
You can also search for this author inPubMed Google Scholar - Schuichi Koizumi
You can also search for this author inPubMed Google Scholar - C. Justin Lee
You can also search for this author inPubMed Google Scholar - Shane A. Liddelow
You can also search for this author inPubMed Google Scholar - Brian A. MacVicar
You can also search for this author inPubMed Google Scholar - Pierre Magistretti
You can also search for this author inPubMed Google Scholar - Albee Messing
You can also search for this author inPubMed Google Scholar - Anusha Mishra
You can also search for this author inPubMed Google Scholar - Anna V. Molofsky
You can also search for this author inPubMed Google Scholar - Keith K. Murai
You can also search for this author inPubMed Google Scholar - Christopher M. Norris
You can also search for this author inPubMed Google Scholar - Seiji Okada
You can also search for this author inPubMed Google Scholar - Stéphane H. R. Oliet
You can also search for this author inPubMed Google Scholar - João F. Oliveira
You can also search for this author inPubMed Google Scholar - Aude Panatier
You can also search for this author inPubMed Google Scholar - Vladimir Parpura
You can also search for this author inPubMed Google Scholar - Marcela Pekna
You can also search for this author inPubMed Google Scholar - Milos Pekny
You can also search for this author inPubMed Google Scholar - Luc Pellerin
You can also search for this author inPubMed Google Scholar - Gertrudis Perea
You can also search for this author inPubMed Google Scholar - Beatriz G. Pérez-Nievas
You can also search for this author inPubMed Google Scholar - Frank W. Pfrieger
You can also search for this author inPubMed Google Scholar - Kira E. Poskanzer
You can also search for this author inPubMed Google Scholar - Francisco J. Quintana
You can also search for this author inPubMed Google Scholar - Richard M. Ransohoff
You can also search for this author inPubMed Google Scholar - Miriam Riquelme-Perez
You can also search for this author inPubMed Google Scholar - Stefanie Robel
You can also search for this author inPubMed Google Scholar - Christine R. Rose
You can also search for this author inPubMed Google Scholar - Jeffrey D. Rothstein
You can also search for this author inPubMed Google Scholar - Nathalie Rouach
You can also search for this author inPubMed Google Scholar - David H. Rowitch
You can also search for this author inPubMed Google Scholar - Alexey Semyanov
You can also search for this author inPubMed Google Scholar - Swetlana Sirko
You can also search for this author inPubMed Google Scholar - Harald Sontheimer
You can also search for this author inPubMed Google Scholar - Raymond A. Swanson
You can also search for this author inPubMed Google Scholar - Javier Vitorica
You can also search for this author inPubMed Google Scholar - Ina-Beate Wanner
You can also search for this author inPubMed Google Scholar - Levi B. Wood
You can also search for this author inPubMed Google Scholar - Jiaqian Wu
You can also search for this author inPubMed Google Scholar - Binhai Zheng
You can also search for this author inPubMed Google Scholar - Eduardo R. Zimmer
You can also search for this author inPubMed Google Scholar - Robert Zorec
You can also search for this author inPubMed Google Scholar - Michael V. Sofroniew
You can also search for this author inPubMed Google Scholar - Alexei Verkhratsky
You can also search for this author inPubMed Google Scholar
Contributions
A.L., A.S.P., A. Verkhratsky, A. Volterra, C.E., C.S., E.G., G.C., G.C.P., J.P.O., and M.V.S. participated in the initial discussion and drafted the outline. C.E. and E.G. prepared the tables, and C.E., E.G., and M.V.S. prepared the figures. A.L., A.S.P., A. Verkhratsky, C.E., C.S., E.G., G.C.P., and J.P.O. wrote parts of the manuscript. E.G. and A. Verkhratsky assembled a joint text with the help of C.E. and M.V.S. The manuscript was then edited by A.L., A.S.P., A. Volterra, C.S., G.C., G.C.P., and J.P.O. The rest of the authors fact-checked, improved accuracy, and provided content that was integrated by C.E., E.G., A. Verkhratsky, and M.V.S., and validated by A.L., A.S.P., A. Volterra, C.S., G.C., G.C.P., and J.P.O. The manuscript was circulated several times among all the authors until no mistakes or inaccuracies were detected and no disagreement was expressed by any author. All authors have approved the final version of the manuscript.
Corresponding authors
Correspondence toCarole Escartin, Elena Galea, Michael V. Sofroniew or Alexei Verkhratsky.
Ethics declarations
Competing interests
C.F. received grants from Teva, Novartis, and Merck-Serono. The rest of the authors declare no conflicts of interest.
Additional information
Peer review information Nature Neuroscience thanks Won-Suk Chung, Frank Heppner, and Ye Zhang for their contribution to the peer review of this work.
Rights and permissions
About this article
Cite this article
Escartin, C., Galea, E., Lakatos, A. et al. Reactive astrocyte nomenclature, definitions, and future directions.Nat Neurosci 24, 312–325 (2021). https://doi.org/10.1038/s41593-020-00783-4
- Received: 13 August 2020
- Accepted: 16 December 2020
- Published: 15 February 2021
- Issue Date: March 2021
- DOI: https://doi.org/10.1038/s41593-020-00783-4