COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer (original) (raw)
Main
Overexpression of cyclooxygenase (COX)-2 is now recognized as a marker for tumour progression documented for cancers of the colon (Soslow et al, 2000), lung (Hida et al, 1998; Soslow et al, 2000), head and neck (Chan et al, 1999), pancreas (Tucker et al, 1999) and the breast (Parrett et al, 1997; Soslow et al, 2000). A functional role of COX-2 in tumour development and progression has been demonstrated by both overexpression (Liu et al, 2001) and disruption (Chulada et al, 2000) of the COX-2 gene as well as application of drugs blocking both COX-1/-2 or COX-2 alone (Lala et al, 1997; Harris, 2003; Wang and DuBois, 2004). This role has primarily been attributed to elevated levels of prostanoids, mainly prostaglandin E2 (PGE2), in the tumour microenvironment (Rolland et al, 1980). We had earlier demonstrated that tumour-derived PGE2 acts as a paracrine as well as an autocrine factor to promote breast cancer progression and metastasis by multiple mechanisms, namely by inactivation of host anti tumour immune cells and a stimulation of tumour cell migration, invasiveness and tumour-associated angiogenesis (Lala and Saarloos, 1994; Lala et al, 1997; Rozic et al, 2001).
PGE2 action depends on activation of one or more of the four PGE2 receptors (EP1-EP4) expressed by target cells. They are encoded by different genes and coupled with different G-proteins: EP1 coupled with Gq, EP2 and EP4 coupled with Gs, and certain transcripts of EP3 coupled with Gi (Breyer et al, 2001). Role(s) of specific EP receptor-mediated signalling in tumour development and progression has so far been shown to vary with the tumour model and the specific cellular functions contributing to the metastatic phenotype of cancer cells. For example, EP1, EP2 and EP4 contributed to colon carcinogenesis (Hull et al, 2004), and EP2 was shown to be required for COX-2-mediated mammary hyperplasia (Chang et al, 2005). Furthermore, EP4 contributed to the stimulation of migration of colorectal (Sheng et al, 2001) and breast (Timoshenko et al, 2003) cancer cells. EP4 receptors were also responsible for an upregulation of iNOS gene expression under inducible conditions in murine breast cancer cells that increased their invasive capacity (Timoshenko et al, 2004), as well as in osteoclast development and bone metastasis in a breast cancer model (Ohshiba et al, 2003).
Whereas the role of COX-2 in promoting tumour-associated angiogenesis is well-documented (Tsujii et al, 1998), possible role of COX-2 in lymphangiogenesis and lymphatic metastasis remains poorly defined. Two members of the vascular endothelial growth factor (VEGF) family that is, VEGF-C and VEGF-D have been shown to promote lymphangiogenesis by binding to VEGF receptor VEGFR-3 on lymphatic endothelial cells (Saharinen et al, 2004). Forced VEGF-C overexpression in a VEGF-C-nonexpessing and nonmetastatic human breast cancer cell line MCF-7 resulted in enhanced tumour growth in vivo, lymphangiogenesis and lymphatic metastasis in immunodeficient mice (Mattila et al, 2002). Elevated expression of VEGF-C in tumour tissues has been shown to have a negative influence on prognosis and a positive correlation with lymph node metastasis in many cancers including cancers of the breast (Nakamura et al, 2003), uterine cervix (Fujimoto et al, 2004), colon and rectum (Onogawa et al, 2004), oesophagus (Kimura et al, 2003), stomach (Duff et al, 2003a), head and neck (O-charoenrat et al, 2001), and gallbladder (Nakashima et al, 2003). Additionally, serum VEGF-C was shown to be elevated in patients with non-small cell lung cancer (Tamura and Ohta, 2003) and colorectal cancer (Duff et al, 2003b), and in the former case, this was also correlated with lymph node metastasis. Interestingly, a positive association between COX-2 and VEGF-C mRNA expression has been reported in oesophageal adenocarcinoma (von Rahden et al, 2005). A similar association between COX-2 and VEGF-C was also demonstrated at the protein levels by immunohistochemical studies of squamous cell carcinomas of the head and neck (Kyzas et al, 2005) and oesophagus (Byeon et al, 2004) as well as in non-small cell lung adenocarcinoma (Su et al, 2004). A role of COX-2 in VEGF-C upregulation was suggested in the case of non-small cell lung cancer cells (Su et al, 2004) as well as oesophageal adenocarcinoma cells (von Rahden et al, 2005). To date, however, no information exists regarding whether COX-2 is causally associated with VEGF-C upregulation and thereby lyphangiogenesis in breast cancer, and if so, what is the role of EP receptors on cancer cells in this event.
Whether cancer metastasis to lymph nodes depends on pre-existing or newly formed lymphatics still remains a debated issue. Intratumoral lymphangiogenesis, as identified by the lymphatic endothelium-specific marker LYVE-1 (Saharinen et al, 2004), is a salient feature of invasive head and neck cancer (Beasley et al, 2002) and also inflammatory breast cancer (Van der Auwera et al, 2004). However, this is not so for noninflammatory, invasive human breast carcinomas (Williams et al, 2003; Vleugel et al, 2004; Van der Auwera et al, 2004). It the latter case, lymphatic vessels were demonstrated only in the peritumoral region (Vleugel et al, 2004) and it is unclear whether they represent pre-existing or newly formed lymphatics. Thus a paracrine role of breast cancer-derived VEGF-C in lymphangiogenesis and lymphatic metastasis remains an open question.
Present study utilized several well-established human breast cancer cell lines varying in COX-2 expression and metastatic abilities as well as numerous human breast cancer specimens with the following objectives: (1) to examine the relationship between COX-2 (mRNA) expression and VEGF-C (mRNA) expression (cell lines and tissues) or VEGF-C secretion (cell lines); (2) to examine the relationship between COX-2 or VEGF-C expression and the expression of LYVE-1, a marker for the lymphatic endothelium, in breast cancer tissues; (3) to examine the causal relationship between COX-2 activity or gene expression and VEGF-C expression/secretion in high COX-2 and VEGF-C expressing cell lines; (4) to identify the role (s) of specific EP receptors in endogenous PGE2-mediated VEGF-C stimulation in these cells.
Materials and methods
Reagents
PGE2, 17-phenyl trinor PGE2 (EP1 agonist) and SC-560 (selective COX-1 inhibitor) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). NS-398 (selective COX-2 inhibitor), PP1 (Src kinase inhibitor) and SC-51322 (EP1 antagonist) were from Biomol (Plymouth Meeting, PA, USA). AH-23848B (EP4 antagonist) was from Glaxo/Wellcome (Stevenage, UK). Concanavalin A (Con A), indomethacin (nonselective COX-1/COX-2 inhibitor), and 3,3′-diaminobenzidine tablets were from Sigma (Oakville, ON, Canada). PD153035 (Her2/neu kinase inhibitor) and SB203580 (p38 kinase inhibitor) were from Calbiochem (San Diego, CA, USA). L-161982 (EP4 antagonist) was kindly provided by Dr M Young from Merck Frosst (Kirkland, QC, Canada).
Human breast cancer cell lines
Human breast cancer cell lines MCF-7, T-47D, Hs578T and MDA-MB-231 were obtained from the American Type Culture Collection and grown in DMEM (Invitrogen/GIBCO, Burlington, ON, Canada) supplemented with 8% FBS, 25 mM HEPES buffer, 50 U ml−1 penicillin and 50 _μ_g ml−1 streptomycin. Propagation of MCF-7 and T-47D cells were performed in the presence of 0.01 mg ml−1 bovine insulin.
Human breast cancer tissues
Frozen tissue samples of 10 surgically resected human breast cancer specimens were obtained from The London Health Sciences Center, London Laboratory Services Group, London, Ontario without any preselection. The study was approved by the Tissue and Archives Committee, Department of Pathology, the University of Western Ontario. Histological data were available on eight out 10 specimens. Of these, two were lymph node positive and six had no demonstrable metastasis in the resected lymph nodes. The tumours represented infiltrating, invasive ductal, lobular, or ducto-lobular carcinomas of various SBR grades (I–III). None was described as inflammatory breast cancer.
RT–PCR
First-strand cDNAs were synthesized from 2 _μ_g of TRIzol reagent-extracted total RNA from breast cancer cells and lesions using the SuperScript™ II Reverse Transcriptase (Invitrogen, Burlington, ON, Canada). Regular hot start (2 min, 94°C) PCR was performed in a 20 _μ_l volume containing 18 _μ_l Platinum® PCR SuperMix (Invitrogen), 0.8 _μ_l template cDNA solution, and 0.8 _μ_l primer mixture (25 pmol _μ_l−1 each). PCR was run for 30–35 cycles of denaturation 94°C (30 s), annealing 55°C (30 s), extension 72°C (45 s) followed by 5 min of final extension at 72°C. Primers for VEGF family, COX-2, LYVE-1 and GAPDH (Table 1) were synthesized locally at the UWO Oligo Factory (London, Canada) or ordered from Sigma/Genosys (Oakville, ON, Canada). All primers were designed and evaluated using Oligo Explorer and Oligo Analyzer software (Teemu Kuulasmaa, Finland) except for LYVE-1, VEGF-A and VEGF-D pairs (41–43). PCR products were separated on 1% agarose gel containing 0.25 _μ_g ml−1 ethidium bromide and visualized under UV light. Real-time quantitative PCR (qPCR) for VEGF-C, COX-2, LYVE-1 and GAPDH was performed in single microcapillary tubes using the LightCycler™ (Roche Diagnostic, Laval, Canada) and SYBR® Green Tag ReadyMix™ (Sigma, St Louis, USA). Cycling parameters were optimized as follow: denaturation 94°C (0 s), annealing 55°C (5 s), extension 72°C (24 s) and detection 80°C (1 s). Each microcapillary contained 7.1 _μ_l nuclease free H2O, 10 _μ_l SYBR reagent, 0.5 _μ_l template cDNA, 1.6 _μ_l 25 mM MgCl2 and 0.8 _μ_l 25 pmol _μ_l−1 primer mixture. The cycler software was used for quantification of COX-2, VEGF-C and LYVE-1 mRNA levels relative to GAPDH mRNA expression.
Table 1 Oligonucleotide Primer Pairs for RT–PCR
ELISA for VEGF-A, VEGF-C and VEGF-D
The levels of VEGF-A and VEGF-C accumulating in serum-free cell culture media were measured using Human VEGF and VEGF-C EIA kits from Immuno-Biological Laboratories (Gumna, Japan). The levels of VEGF-D were measured using Quantikine® Human VEGF-D Immunoassay kit from R&D (Minneapolis, MN, USA).
COX-2 siRNA transfection
The Silencer siRNA Transfection Kit and predesigned siRNA from Ambion (St Austin, TX, USA) were used to transfect MDA-MB-231 cells with COX-2 siRNA by neofection method. The conditions of neofection were optimized by using GAPDH siRNA as a positive control and siPORT NeoFX was selected as the most efficient transfection agent. To perform transfection with COX-2 siRNA, 2.3 ml of cells (0.1 × 106 cells ml−1) in complete DMEM were mixed with 200 _μ_l of the transfection complex containing 125 nM of siRNA and 2% of siPORT NeoFX in OPTI-MEM I medium and added to a well of six-well culture plate. The cells were cultured for 48 h at 37°C and the efficiency of transfection was assayed by qPCR. To analyse VEGF-C secretion, the monolayer of COX-2 siRNA-treated cells was rinsed with serum-free DMEM and incubated for additional 24 h in the serum-free medium (2 ml well−1).
Immunohistochemistry for VEGF-C
MDA-MB-231 cells were grown up to subconfluency on Lab-Tek Permanox slides with four chambers from Nalge Nunc (Naperville, IL, USA). The complete medium was replaced by serum-free DMEM and the cells were preincubated for 1 h with serum-free DMEM followed by 24 h incubation with or without inhibitors as specified in the results. The cell monolayers were rinsed with PBS, fixed in 2% formaldehyde for 30 min, washed again two times with PBS, and treated for 5 min with 2% glycine. The slides were then immunostained using anti-human VEGF-C rabbit IgG from IBL (Gunma, Japan), the Vestatin Elite ABC kit from Vector Laboratories (Burlingame, CA, USA) and 3,3′-diaminobenzidine for colour development according to the manufacturers'protocol.
MTT assay for cell proliferation/survival
All agents (pharmacological agents, inhibitors), the effects of which were tested on VEGF-C production by human breast cancer cells were also tested under identical conditions for possible effects on cell proliferation/survival using the MTT Cell Proliferation Kit I from Roche Diagnostics (Laval, QC, Canada), as reported earlier (Timoshenko et al, 2003).
Statistics
All mean values and standard deviations (s.d.) from at least triplicate (often quadruplicate) measurements were calculated with Microsoft Office Excel 2003 (Microsoft Corporation, Seattle, WA, USA). Statistical significance of differences between two groups was determined with a two-sided Student's _t_-test considering P<0.05 as an indicator of significant difference between means. Correlations between levels of COX-2, VEGF-C and LYVE-1 mRNA levels were made with nonparametric Spearman correlation test providing correlation coefficients and _P_-values. Linear regression analysis of the data including 95% confidence intervals was performed with statistical package of GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
Results
High VEGF-C expression and production distinguishes a highly metastatic from a nonmetastatic breast cancer cell line
Highly metastatic MDA-MB-231 and nonmetastatic MCF-7 human breast cancer cell lines were found to express the mRNA, although at different levels, for all four VEGFs (A, B, C, D) as detected by RT–PCR (Figure 1A). Both cell lines secreted immunodetectable levels of VEGF-A and VEGF-C but very little VEGF-D at 24 h (Figure 1B). MCF-7 cells produced low levels of VEGF-C as well as VEGF-A. In comparison with MCF-7 cells, VEGF-A production by MDA-MB-231 cells was two-fold higher, whereas VEGF-C production was about 25-fold higher (Figure 1B). These findings are consistent with overexpression of VEGF-C mRNA in MDA-MB-231 cells as opposed to low expression in MCF-7 cells, as measured by real-time qPCR (Figure 1C). Accumulation of both VEGF-A and VEGF-C in serum-free culture media of MDA-MB-231 cells increased linearly for at least 48 h and, again, no detectable level of VEGF-D was noted at any time point (Figure 1D). The rate of VEGF-C production by these cells was about 10-fold higher than that of VEGF-A. Thus, the elevated expression and production of the lymphangiogenic factor VEGF-C seems to be an important feature of this highly metastatic breast cancer cell line.
Figure 1
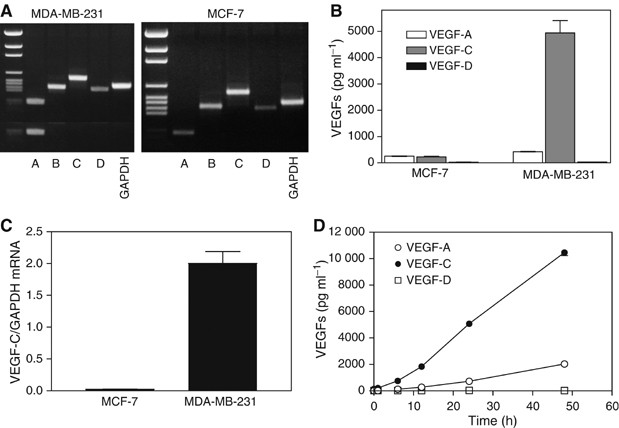
Expression of VEGF-A, -B, -C and -D mRNAs and VEGF-A, -C, and –D protein secretion by human breast cancer cell lines MDA-MB-231 and MCF-7. (A) RT–PCR-based amplification of VEGF mRNAs, showing that message for each VEGF class is detectable in both cell lines. (B) Accumulation of secreted VEGF-A, -C and -D proteins in serum-free culture media of both cell lines at 24 h showing high VEGF-C production by MDA-MB-231 cells. (C) Differences in expression of VEGF-C mRNA by MDA-MB-231 and MCF-7 cells as revealed by real time qPCR. (D) Kinetics of accumulation of VEGF-A, -C and -D protein in culture media of MDA-MB-231 cells. Data in (B, C, and D) represent mean±s.d. (_n_=4).
VEGF-C production by human breast cancer cells and VEGF-C expression by human breast cancer tissues correlate positively with COX-2 expression
As high levels of COX-2 expressed by MDA-MB-231 cells was shown to contribute to its metastatic phenotype whereas nonmetastatic MCF-7 cells did not express COX-2 (Liu and Rose, 1996), we examined whether there is a relationship between COX-2 expression and VEGF-C producing ability, using additional human breast cancer cell lines T-47D (nonmetastatic) and Hs578T (metastatic).
As detected by RT–PCR, all the four human breast cancer cell lines tested expressed similar levels of COX-1 mRNA but significantly different levels of COX-2 mRNA. Thus, MCF-7 cells were COX-2 negative, T-47D expressed very low levels of COX-2, Hs578T expressed moderately high levels, whereas MDA-MB-231 expressed very high levels of COX-2 (Figure 2A). The levels of VEGF-C secretion by these cell lines at 24 h (Figure 2B) clearly correlated with their relative COX-2 expression levels.
Figure 2
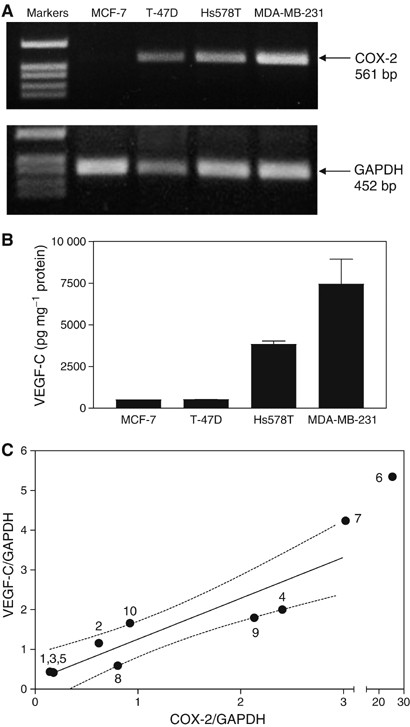
Relationship between COX-2 expression and VEGF-C synthesis/expression in human breast cancer cell lines and tissues. (A) Expression of COX-2 and GAPDH genes in MCF-7, T-47D, Hs578T and MDA-MB-231 human breast cancer cells as detected by RT–PCR. COX-2 expression is not detectable in MCF-7, weak in T-47D, moderate in Hs578T, and high in MDA-MB-231 cells. (B) Accumulation of VEGF-C in culture media of the above breast cancer cell lines at a 24 h correlates with their levels of COX-2 expression. Data represent mean±s.d. (_n_=4). (C) Correlation between relative levels of COX-2 mRNA and VEGF-C mRNA (standardized against GAPDH mRNA) expression in human breast cancer tissues (_n_=10) measured with real-time qPCR reveals a strong positive association between the two parameters (_r_=0.94, _P_=0.0002). Numbers on the graph present patient identifiers, and dotted lines present 95% confidence intervals of the regression line.
To find out whether the observed positive correlation between COX-2 and VEGF-C expression or production by breast cancer cell lines reflect a similar relationship in vivo, we analysed mRNA levels for both genes in human breast cancer tissue samples from 10 randomly selected surgically removed specimens. The results of the real-time qPCR study showed that, indeed, there was a strong positive association (_r_=0.94, _P_=0.0002) between COX-2 and VEGF-C mRNA levels (Figure 2C). Taken together, these data clearly demonstrated a positive association between COX-2 and VEGF-C systems in breast cancer cells in vitro as well as in vivo.
LYVE-1 expression in human breast cancer tissues is positively correlated with COX-2 and VEGF-C expression
LYVE-1 is a highly selective marker of lymphatic endothelial cells (Saharinen et al, 2004) and is not expressed by MDA-MB-231 breast cancer cells (Cunnick et al, 2001). The level of LYVE-1 mRNA expression in tumour samples was shown to be a sensitive indicator of the level of lymphangiogenesis in vivo (Cunnick et al, 2001; Van der Auwera et al, 2004). For this reason, we analysed mRNA level of the LYVE-1 gene in the same breast cancer tissue samples in which COX-2 and VEGF-C mRNA levels were measured. The results of the real time qPCR revealed a robust correlation of LYVE-1 mRNA levels in these tissue samples with COX-2 (_r_=0.75, _P_=0.017) as well as VEGF-C (_r_=0.78, _P_=0.013) expression levels (Figure 3A and B).
Figure 3
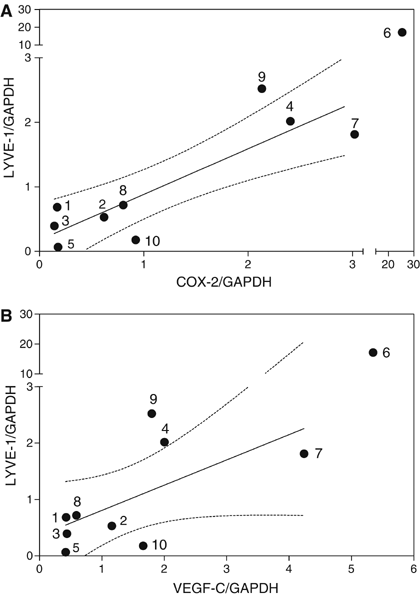
Relationship between LYVE-1 and COX-2 (A) or VEGF-C (B) mRNA expression in the same breast cancer tissues (_n_=10) used for data in Figure 2C. A positive association (_r_=0.75, _P_=0.017 and _r_=0.78, _P_=0.013, respectively) is revealed in both cases; dotted lines present 95% confidence interval of the regression line.
COX-2 activity and VEGF-C expression/production are causally related
To examine whether COX-2 activity played any role in regulating VEGF-C synthesis by human breast cancer cells, we tested the effects of COX-1/-2 inhibitors (indomethacin, NS-398 and SC-560) on VEGF-C accumulation in cell culture media. For a comparison, VEGF-A production was measured side by side. The inhibitors were added at concentrations which were nontoxic (having no effects on cell proliferation/survival) for cells as detected by MTT assay (data not shown). We found that the nonselective COX-1/-2 inhibitor indomethacin (20 μ M) as well as the selective COX-2 inhibitor NS-398 (50 μ M) strongly suppressed but did not abrogate VEGF-C production by both COX-2 expressing cell lines MDA-MB-231 (Figure 4A) and Hs578T (Figure 4B). Similar inhibitory effects of COX-1/-2 and COX-2 inhibitors implicate the regulatory role of COX-2 on VEGF-C production. In contrast, VEGF-A production by MDA-MB-231 cells was not reduced in the presence of these inhibitors (Figure 4A). We had earlier shown that these concentrations of indomethacin and NS-398, respectively, suppressed PGE2 production by 72 and 94% in MDA-MB-231 cells (Timoshenko et al, 2003). Concanavalin A (Con A), earlier shown to stimulate PGE2 production by MDA-MB-231 cells (Timoshenko et al, 2003), also stimulated VEGF-C as well as VEGF-A production, which were significantly blocked with the COX-1/2 inhibitor (Figure 4A). This inhibition was much higher in the case of VEGF-C. A small inhibitory effect on VEGF-C but not VEGF-A was also observed with the selective COX-1 inhibitor SC-560 (5 μ M) (Figure 4A and B) but it was significantly less than that caused by the selective COX-2 inhibitor NS-398 (Figure 4A). In line with these findings, NS-398 and indomethacin were also found to downregulate VEGF-C mRNA expression in MDA-MB-231 cells validating a role of COX-2 as an upstream regulatory enzyme (Figure 4C). These results, taken together, reveal that VEGF-C synthesis by breast cancer cells is, at least in part, upregulated by endogenous COX-2 activity.
Figure 4
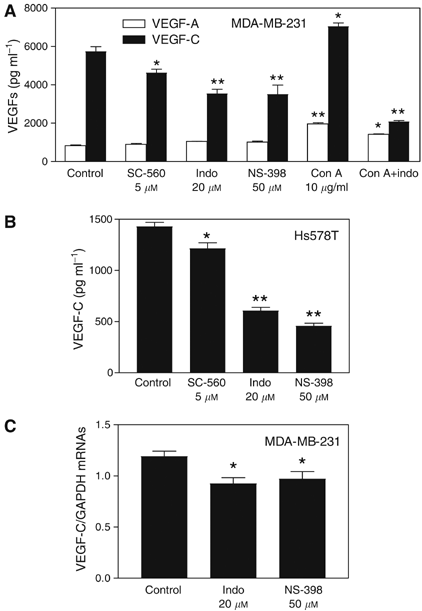
Effects of COX inhibitors on VEGF-C and VEGF-A secretion and VEGF-C expression in breast cancer cells. (A) Effects of COX-1 inhibitor SC-560, COX-1/-2 inhibitor indomethacin, and COX-2 inhibitor NS-398 on VEGF-C or VEGF-A production by COX-2 expressing MDA-MB-231 cells. (B) Effects of same inhibitors on VEGF-C production by COX-2 expressing Hs578T cells. COX-1 inhibitor caused a minor (but significant; P<0.05) reduction, whereas COX-2 or COX-1/-2 inhibitors caused a substantial (P<0.01) and similar reduction in VEGF-C production by both cell lines. However, these inhibitors have no inhibitory effect on VEGF-A production. Con A, known to stimulate PGE2 production by MDA-MB-231 cells, also stimulated VEGF-C as well as VEGF-A production, which were substantially (P<0.01) blocked with COX-1/2 inhibitor. (C) Effect of 24 h treatment with COX inhibitors (20 μ M indomethacin or 50 μ M NS-398 in serum-free culture) on VEGF-C mRNA expression in MDA-MB-231 cells. Data represent mean±s.d. (_n_=4). *P<0.05 and **P<0.001 in comparison to control untreated cells.
Knock down of COX-2 mRNA reduces VEGF-C production
To examine whether the COX-2 gene plays a regulatory role in VEGF-C synthesis by human breast cancer cells, we adopted the siRNA approach to knock down COX-2 gene in high COX-2 expressing MDA-MB-231 cells. As shown in Figure 5, COX-2 siRNA-treated cells exhibited a significant reduction in both COX-2 mRNA expression as well as VEGF-C production. However, the level of reduction in VEGF-C production by cells during a 24 h period following COX-2 siRNA pretreatment for 48 h was relatively less then the levels of reduction in COX-2 mRNA expression either at 48 or 72 h. These data may indicate that the presence of additional gene (s) other than COX-2 regulating VEGF-C synthesis in MDA-MB-231 cells.
Figure 5
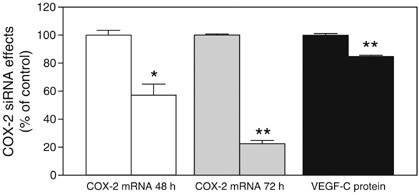
Effects of COX-2 siRNA treatment on COX-2 expression and VEGF-C production by MDA-MB-231 cells. The first bar in each series is control, that is, the treatment with scrambled siRNA, and the second bar is experimental, that is, the treatment with COX-2 siRNA. siRNA treatment was conducted in serum containing medium for 48 h. Cells were washed and cultured in serum free media for another 24 h to measure VEGF-C protein accumulation in the cell free media, and COX-2 mRNA levels in cells at both 48 and 72 h following the siRNA treatment. A significant suppression of COX-2 mRNA expression as well as VEGF-C secretion was noted in COX-2 siRNA treated cells. *P<0.01, **P<0.001.
EP1 and EP4 receptors contribute to VEGF-C production by highly metastatic human breast cancer cells
We had earlier shown that PGE2 is the major prostanoid resulting from COX-2 expression in highly metastatic breast cancer cells of different origin and that MDA-MB-231 cells express mRNA for each of the four PGE2 receptors (Timoshenko et al, 2003). We had also shown that EP4 receptors, which are coupled to Gs proteins, are functional in MDA-MB-231 cells, contributing to their migration in response to endogenous PGE2 (Timoshenko et al, 2003). EP1 receptors are coupled with Gq proteins and typically they activate Ca2+-dependent intracellular signalling cascades. We demonstrated the functionality of EP1 receptors in MDA-MB-231 cells from a transient and moderate increase in intracellular Ca2+ levels in response to exogenous PGE2 (10 μ M) and an EP1 receptor agonist 17-phenyl trinor PGE2 (10 μ M) (not shown).
To test the role of individual EP receptors in an autocrine, PGE2-mediated regulation of VEGF-C synthesis, we treated COX-2-expressing Hs578 T and MDA-MB-231 cells with several EP antagonists including SC-51322, AH-23848B, and L-161982 at nontoxic final concentrations (10, 10 and 1 μ M, respectively) having no effect on cell proliferation/survival (data not shown) but earlier shown to block receptor activity (Coleman et al, 1994a, 1994b; Breyer et al, 2001; Tomita et al, 2002; Timoshenko et al, 2003). A strong inhibition of VEGF-C secretion was found with the selective EP1 receptor antagonist SC-51322 for both cell lines (Figure 6A and B). Two EP4 receptor antagonists, AH-23848B and L-161982, also variably inhibited VEGF-C secretion, however, the effects of L-161982 were stronger than those of AH-23848B. This difference is explained by a higher specificity and affinity of the former compound for EP4 receptors than the latter, which has a crossreactivity with TP receptors (Coleman et al, 1994a, 1994b). Inhibition of VEGF-C secretion in the presence of EP1/EP4 antagonists was at least partially due to downregulation of VEGF-C gene expression as demonstrated by real-time qPCR with MDA-MB-231 cells (Figure 6C). Possible role of EP2 and EP3 receptors could not be tested due to nonavailability of highly selective antagonists.
Figure 6
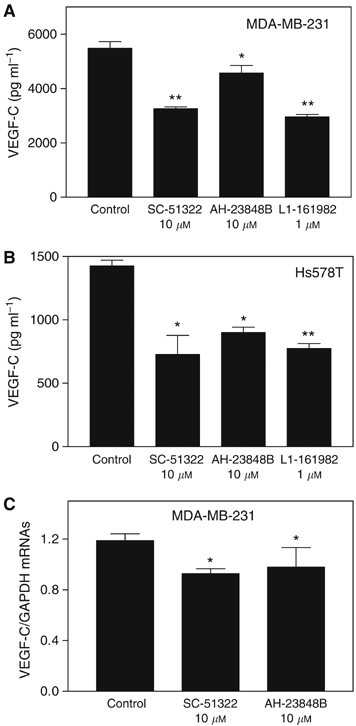
Roles of EP receptors in the regulation of VEGF-C production and expression by COX-2 expressing human breast cancer cells. Cells were treated for 24 h with antagonists of EP1 receptors (SC-51322) and EP4 receptors (AH-23848B and L-161982) in serum-free DMEM. (A and B) Effects of EP receptor antagonists on VEGF-C accumulation in culture media of MDA-MB-231 cells (A) and Hs538T cells (B). A consistent and significant inhibition of VEGF-C production was observed with highly specific EP1 receptor antagonist SC-51322 and EP4 receptor antagonist L-161982. (C) Quantification of VEGF-C mRNA levels in MDA-MD-231 cells after the 24 h exposure to EP receptor antagonists. Data represent mean±s.d. (_n_=4). *P<0.05, **P<0.01.
VEGF-C synthesis by MDA-MB-231 cells is inhibited by inhibitors of Her-2/neu, Src and p38 MAP kinases
VEGF-C synthesis in other cell types was reported to utilize signalling pathways associated with Her2/neu, Src, and p38 MAP kinases (Tsai et al, 2003; Su et al, 2004). We tested whether the application of the respective kinase inhibitors affected VEGF-C synthesis by MDA-MB-231 breast cancer cells. Cells were treated with PD153035 (Her2/neu kinase inhibitor; 5 μ M), PP1 (Src kinase inhibitor, 10 μ M) and SB203580 (p38 MAP kinase inhibitor, 30 μ M). As shown in Figure 7, all these inhibitors at nontoxic concentrations (having no effect on cell proliferation/survival as revealed by the MTT assay) inhibited VEGF-C secretion as well as the level of immunostaining for cytoplasmic VEGF-C production.
Figure 7
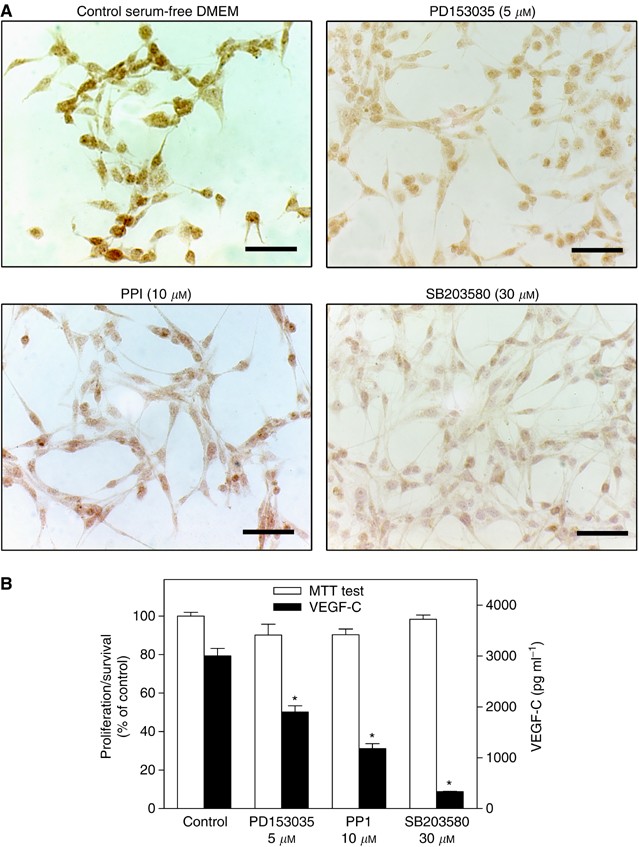
Reduction of VEGF-C expression in MDA-MB-231 cells induced by kinase inhibitors for Her2/Neu (PD153035), Src (PP1) and p38 MAPK (SB203580). MDA-MB-231 cells were cultured for 24 h in serum-free DMEM supplemented with 0.2 % BSA. (A) imunohistochemistry; all the kinase inhibitors at the concentrations employed inhibited cytoplasmic immunostaining for VEGF-C. Scale bar represents 60 _μ_m. (B) cell proliferation/survival (MTT test) and VEGF-C secretion; VEGF-C secretion, but not proliferation/survival, was inhibited by all the inhibitors. Data represent mean±s.d. (_n_=4), *P<0.001.
Discussion
The present study demonstrates for the first time that mRNA levels of COX-2, a well recognized functional marker for tumour progression, are highly correlated with VEGF-C mRNA levels in human breast cancer tissues and VEGF-C gene expression or secretion by breast cancer cell lines; that COX-2 or VEGF-C mRNA expression levels in breast cancer tissues are correlated with the expression of LYVE-1, a marker for lymphangiogenesis; that VEGF-C synthesis in breast cancer cells is stimulated, at least in part, by COX-2, EP1 and EP4 receptor activity.
The stimulatory role of COX-2 in breast cancer progression has earlier been explained by multiple PGE2-dependent mechanisms: an inactivation of antitumour immune cells (Lala and Saarloos, 1994), a stimulation of cancer cell growth and survival (Basu et al, 2005), migration (Rozic et al, 2001; Timoshenko et al, 2003), invasiveness (Rozic et al, 2001) and angiogenesis (Lala et al, 1997; Rozic et al, 2001). Present study demonstrates an additional role of COX-2-in human breast cancer: a stimulation of VEGF-C and thereby lymphangiogenesis in situ, also reported recently for non-small cell lung cancer cells (Su et al, 2004). The role of lymphangiogenesis for lymphatic metastases in human breast cancer patients remains a controversial issue. Intratumoural lymphangiogenesis is a feature of inflammatory breast cancer (Van der Auwera et al, 2004), whereas peritumoral but not intratumoral lymphatics were demonstrated in invasive, noninflammatory breast cancer (Vleugel et al, 2004). In the present study, which measured LYVE-1 mRNA levels in noninflammatory breast cancer tissues as indicators of lymphangiogenesis, the location of the lymphatics remains undetermined. In situ hybridization and immunostaining on a larger number of samples remain as future goals to resolve this issue. It is interesting to note that VEGF-C immunostaining in breast cancer tissues was reported to show a significant correlation with tumour cell invasion of lymphatic vessels at the microscopic level, but not with lymph node metastasis in one study (Kinoshita et al, 2001), whereas another study using a larger sample size including a higher proportion of VEGF-C expressing specimens demonstrated a positive correlation with nodal metastasis (Nakamura et al, 2003). Further studies are needed to examine whether lymphangiogenic role of VEGF-C is essential for lymphatic metastasis of breast cancer. Nevertheless, VEGF-C overexpression can now be added to the list of biomarkers such as overexpression of COX-2, Her-2/neu and VEGF-A which indicate poor prognosis in breast cancer patients (Zhang et al, 2003).
A strong positive association between COX-2 and VEGF-C expression noted here in breast cancer cell lines as well as in breast cancer tissues would suggest that breast cancer cells within the lesions served as the source of COX-2 or VEGF-C. However, we have not excluded the possibility that stromal cells and/or immigrant leukocytes may also be the source of both molecules. COX-2 mRNA expression has also been positively correlated with VEGF-A mRNA expression in human breast cancer specimens (Kirkpatrick et al, 2002), however, this association is much weaker than that we have noted with VEGF-C (correlation coefficients 0.55 vs 0.94). An association between COX-2 and VEGF-C, either at the mRNA or protein levels, has also been reported for squamous cell carcinomas of the head and neck (Kyzas et al, 2005), oesophagus (Byeon et al, 2004; von Rahden et al, 2005), and non-small cell lung cancer (Su et al, 2004). This relationship can be explained in two ways: that both genes are upregulated by a common factor, or that one upregulates the other. The first explanation derives support from the facts that certain growth factors and inflammatory cytokines (such as IL-1_β_, TNF_α_, PDGF, TGF_β_ and heregulin-_β_1) can stimulate VEGF-C mRNA expression or protein synthesis in certain cells (Enholm et al, 1997; Ristimäki et al, 1998; Tsai et al, 2003), and that they can also upregulate COX-2 which is a cytokine-responsive gene (Ristimäki et al, 1994). We have not excluded this possibility in situ. The second explanation, that is, COX-2-mediated upregulation of VEGF-C has been validated in the present study using breast cancer cell lines and was also reported with cell lines derived from non-small cell lung cancer (Su et al, 2004) as well as oesophageal adenocarcinoma (von Rahden et al, 2005). However, our data show that COX-2 is an important, but not the sole regulator of VEGF-C, since inhibition of COX-2 activity or a knock down of the COX-2 gene caused a moderate but not absolute suppression of VEGF-C expression and secretion. The existence of NF-_κ_B binding sites in the promoter regions of both genes (Appleby et al, 1994; Chilov et al, 1997) may suggest additional intrinsic mediator(s) causing a parallel upregulation of both genes via NF-_κ_B pathway.
We have shown that COX-2-mediated upregulation of VEGF-C is, at least in part, dependent on endogenous PGE2-mediated signalling via EP1 and EP4 receptors. EP1 activation was also reported to contribute to VEGF-C upregulation in non-small cell lung cancer cells (Su et al, 2004). We had earlier reported the contribution of EP4 in endogenous PGE2-stimulated migration of MDA-MB-231 cells (Timoshenko et al, 2003), but did not exclude the role of EP1 in this process. EP2 has recently been implicated in COX-2-mediated mammary hyperplasia (Chang et al, 2005). Taken together, these results reveal that EP1, EP2 and EP4 receptors contribute to breast cancer progression, similar to their documented roles in experimental colon carcinogenesis (Hull et al, 2004).
Downstream signalling molecules responsible for EP1- or EP4-mediated VEGF-C upregulation in breast cancer remain to be identified. The promoter region of VEGF-C gene contains putative binding sites for Sp1, AP-2 and NF-_κ_B (Chilov et al, 1997) and, therefore, activation of any of these transcription factors may be instrumental in upregulation of VEGF-C. VEGF-C upregulation in case of non-small cell lung cancer cells was shown to follow EP1-mediated transactivation of Her-2/neu via Src kinase pathway (Su et al, 2004). In turn, Src kinase pathway, in some systems, was reported to cause activation of NF-_κ_B (Courter et al, 2005) or Sp1 (Xu et al, 2004). Furthemore, Her-2/neu kinase stimulation by heregulin-_β_1 was shown to upregulate VEGF-C in COX-2 negative MCF-7 cells following activation of p38 MAP kinase and NF-_κ_B (Tsai et al, 2003). In support of some of these findings, we have shown here that VEGF-C synthesis by COX-2 expressing MDA-MB-231 breast cancer cells was dependent on Her-2/neu, p38 MAP and Src kinases. Whether and how these pathways are triggered by activation of EP1 or EP4 in breast cancer cells remain to be examined. Such studies may highlight newer therapeutic targets for breast cancer in addition to COX-2, Her2/neu and EP receptors as revealed here.
Accession codes
Accessions
GenBank/EMBL/DDBJ
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
- Appleby SB, Ristimäki A, Neilson K, Narko K, Hla T (1994) Structure of the human cyclo-oxygenase-2 gene. Biochem J 302: 723–727
Article CAS PubMed PubMed Central Google Scholar - Basu GD, Pathangey LB, Tinder TL, Gendler SJ, Mukherjee P (2005) Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells. Breast Cancer Res 7: R422–R435
Article CAS PubMed PubMed Central Google Scholar - Beasley NJ, Prevo R, Banerji S, Leek RD, Moore J, van Trappen P, Cox G, Harris AL, Jackson DG (2002) Intratumoral lymphangiogenesis and lymph node metastasis in head and neck cancer. Cancer Res 62: 1315–1320
CAS PubMed Google Scholar - Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001) Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41: 661–690
Article CAS PubMed Google Scholar - Byeon JS, Jung HY, Lee YJ, Lee D, Lee GH, Myung SJ, Yang SK, Hong WS, Kim JH, Min YI, Kim JS (2004) Clinicopathological significance of vascular endothelial growth factor-C and cyclooxygenase-2 in esophageal squamous cell carcinoma. J Gastroenterol Hepatol 19: 648–654
Article CAS PubMed Google Scholar - Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL, Dannenberg AJ. (1999) Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res 59: 991–994
CAS PubMed Google Scholar - Chang SH, Ai Y, Breyer RM, Lane TF, Hla T (2005) The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res 65: 4496–4499
Article CAS PubMed Google Scholar - Chilov D, Kukk E, Taira S, Jeltsch M, Kaukonen J, Palotie A, Joukov V, Alitalo K (1997) Genomic organization of human and mouse genes for vascular endothelial growth factor C. J Biol Chem 272: 25176–25183
Article CAS PubMed Google Scholar - Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R (2000) Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res 60: 4705–4708
CAS PubMed Google Scholar - Coleman RA, Grix SP, Head SA, Louttit JB, Mallett A, Sheldrick RL (1994a) A novel inhibitory prostanoid receptor in piglet saphenous vein. Prostaglandins 47: 151–168
Article CAS PubMed Google Scholar - Coleman RA, Smith WL, Narumiya S (1994b) VIII. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46: 205–229
CAS PubMed Google Scholar - Courter DL, Lomas L, Scatena M, Giachelli CM (2005) Src kinase activity is required for integrin _α_V_β_3-mediated activation of nuclear factor-_κ_B. J Biol Chem 280: 12145–12151
Article CAS PubMed Google Scholar - Cunnick GH, Jiang WG, Gomez KF, Mansel RE (2001) Lymphangiogenesis quantification using quantitative PCR and breast cancer as a model. Biochem Biophys Res Commun 288: 1043–1046
Article CAS PubMed Google Scholar - Duff SE, Li C, Jeziorska M, Kumar S, Saunders MP, Sherlock D, O'Dwyer ST, Jayson GC (2003a) Vascular endothelial growth factors C and D and lymphangiogenesis in gastrointestinal tract malignancy. Br J Cancer 89: 426–430
Article CAS PubMed PubMed Central Google Scholar - Duff SE, Li C, Renehan A, O'Dwyer ST, Kumar S (2003b) Immunodetection and molecular forms of plasma vascular endothelial growth factor-C. Int J Oncol 22: 339–343
CAS PubMed Google Scholar - Enholm B, Paavonen K, Ristimäki A, Kumar V, Gunji Y, Klefstrom J, Kivinen L, Laiho M, Olofsson B, Joukov V, Eriksson U, Alitalo K (1997) Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 14: 2475–2483
Article CAS PubMed Google Scholar - Fujimoto J, Toyoki H, Sato E, Sakaguchi H, Tamaya T (2004) Clinical implication of expression of vascular endothelial growth factor-C in metastatic lymph nodes of uterine cervical cancers. Br J Cancer 91: 466–469
Article CAS PubMed PubMed Central Google Scholar - Harris RE (2003) COX-2 blockage in cancer prevention and therapy. New Jersey: Humana Press
Google Scholar - Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T, Takahashi T (1998) Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res 58: 3761–3764
CAS PubMed Google Scholar - Hull MA, Ko SCW, Hawcroft G (2004) Prostaglandin EP receptors. Targets for treatment and prevention of colon cancer? Mol Cancer Ther 3: 1031–1039
Article CAS PubMed Google Scholar - Kimura Y, Watanabe M, Ohga T, Saeki H, Kakeji Y, Baba H, Maehara Y (2003) Vascular endothelial growth factor C expression correlates with lymphatic involvement and poor prognosis in patients with esophageal squamous cell carcinoma. Oncol Rep 10: 1747–1751
PubMed Google Scholar - Kinoshita J, Kitamura K, Kabashima A, Saeki H, Tanaka S, Sugimachi K (2001) Clinical significance of vascular endothelial growth factor-C (VEGF-C) in breast cancer. Breast Cancer Res Treat 66: 159–164
Article CAS PubMed Google Scholar - Kirkpatrick K, Ogunkolade W, Elkak A, Bustin S, Jenkins P, Ghilchik M, Mokbel K (2002) The mRNA expression of cyclo-oxygenase-2 (COX-2) and vascular endothelial growth factor (VEGF) in human breast cancer. Curr Med Res Opin 18: 237–241
Article CAS PubMed Google Scholar - Kyzas PA, Stefanou D, Agnantis NJ (2005) COX-2 expression correlates with VEGF-C and lymph node metastases in patients with head and neck squamous cell carcinoma. Mod Pathol 18: 153–160
Article CAS PubMed Google Scholar - Lala PK, Al-Mutter N, Orucevic A (1997) Effects of chronic indomethacin therapy on the development and progression of spontaneous mammary tumors in C3H/HEJ mice. Int J Cancer 73: 371–380
Article CAS PubMed Google Scholar - Lala PK, Saarloos MN (1994) Prostaglandins and the host immune system: application of prostaglandin inhibitors for cancer immunotherapy. In Prostaglandin inhibitors in tumor immunology and immunotherapy Harris JE, Braun DP, Anderson KM (eds) pp 187–227. Boca Raton: CRC Press
Google Scholar - Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T (2001) Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem 276: 18563–18569
Article CAS PubMed Google Scholar - Liu XH, Rose DP (1996) Differential expression and regulation of cyclooxygenase-1 and -2 in two human breast cancer cell lines. Cancer Res 56: 5125–5127
CAS PubMed Google Scholar - Mattila MMT, Ruohola JK, Karpanen T, Jackson DG, Alitalo K, Härkönen PL (2002) VEGF-C induced lymphangiogenesis is associated with lymph node metastasis in orthotopic MCF-7 tumors. Int J Cancer 98: 946–951
Article CAS PubMed Google Scholar - Nakamura Y, Yasuoka H, Tsujimoto M, Yang Q, Tsukiyama A, Imabun S, Nakahara M, Nakao K, Nakamura M, Mori I, Kakudo K (2003) Clinicopathological significance of vascular endothelial growth factor-C in breast carcinoma with long-term follow-up. Mod Pathol 16: 309–314
Article PubMed Google Scholar - Nakashima T, Kondoh S, Kitoh H, Ozawa H, Okita S, Harada T, Shiraishi K, Ryozawa S, Okita K (2003) Vascular endothelial growth factor-C expression in human gallbladder cancer and its relationship to lymph node metastasis. Int J Mol Med 11: 33–39
CAS PubMed Google Scholar - O-charoenrat P, Rhys-Evans P, Eccles SA (2001) Expression of vascular endothelial growth factor family members in head and neck squamous cell carcinoma correlates with lymph node metastasis. Cancer 92: 556–568
Article CAS PubMed Google Scholar - Ohshiba T, Miyaura C, Ito A (2003) Role of prostaglandin E produced by osteoblasts in osteolysis due to bone metastasis. Biochem Biophys Res Commun 300: 957–964
Article CAS PubMed Google Scholar - Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K (2004) Expression of VEGF-C and VEGF-D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci 95: 32–39
Article CAS PubMed Google Scholar - Parrett ML, Harris RE, Jorder FA, Ross MS, Clausen KP, Robertson FK (1997) Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol 10: 503–507
CAS PubMed Google Scholar - Ristimäki A, Garfinkel S, Wessendorf J, Maciag T, Hla T (1994) Induction of cyclooxygenase-2 by interleukin-1_α_. Evidence for post-transcriptional regulation. J Biol Chem 269: 11769–11775
PubMed Google Scholar - Ristimäki A, Narko K, Enholm B, Joukov V, Alitalo K (1998) Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem 273: 8413–8418
Article PubMed Google Scholar - Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M (1980) Prostaglandin in human breast cancer: Evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J Natl Cancer Inst 64: 1061–1070
CAS PubMed Google Scholar - Rozic JG, Chakraborty C, Lala PK (2001) Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer 93: 497–506
Article CAS PubMed Google Scholar - Saharinen P, Tammela T, Karkkainen MJ, Alitalo K (2004) Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends Immunol 25: 387–395
Article CAS PubMed Google Scholar - Sheng H, Shao J, Washington MK, DuBois RN (2001) Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem 276: 18075–18081
Article CAS PubMed Google Scholar - Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT (2000) COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 89: 2637–2645
Article CAS PubMed Google Scholar - Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC, Kuo ML (2004) Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res 64: 554–564
Article CAS PubMed Google Scholar - Tamura M, Ohta Y (2003) Serum vascular endothelial growth factor-C level in patients with primary non-small cell lung carcinoma: a possible diagnostic tool for lymph node metastasis. Cancer 98: 1217–1222
Article CAS PubMed Google Scholar - Timoshenko AV, Lala PK, Chakraborty C (2004) PGE2-mediated upregulation of iNOS in murine breast cancer cells through the activation of EP4 receptors. Int J Cancer 108: 384–389
Article CAS PubMed Google Scholar - Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C (2003) Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res 289: 265–274
Article CAS PubMed Google Scholar - Tomita M, Li X, Okada Y, Woodiel FN, Young RN, Pilbeam CC, Raisz LG (2002) Effects of selective prostaglandin EP4 receptor antagonist on osteoclast formation and bone resorption in vitro. Bone 30: 159–163
Article CAS PubMed Google Scholar - Tsai PW, Shiah SG, Lin MT, Wu CW, Kuo ML (2003) Up-regulation of vascular endothelial growth factor C in breast cancer cells by heregulin-_β_1. A critical role of p38/nuclear factor-_κ_B signaling pathway. J Biol Chem 278: 5750–5759
Article CAS PubMed Google Scholar - Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN (1998) Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 93: 705–716
Article CAS PubMed Google Scholar - Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT, Fahey 3rd TJ (1999) Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res 59: 987–990
CAS PubMed Google Scholar - Van der Auwera I, Van Laere SJ, Van den Eynden GG, Benoy I, van Dam P, Colpaert CG, Fox SB, Turley H, Harris AL, Van Marck EA, Vermeulen PB, Dirix LY (2004) Increased angiogenesis and lymphangiogenesis in inflammatory versus noninflammatory breast cancer by real-time reverse transcriptase-PCR gene expression quantification. Clin Cancer Res 10: 7965–7971
Article CAS PubMed Google Scholar - Vleugel MM, Bos R, van der Groep P, Greijer AE, Shvarts A, Stel HV, van der Wall E, van Diest PJ (2004) Lack of lymphangiogenesis during breast carcinogenesis. J Clin Pathol 57: 746–751
Article CAS PubMed PubMed Central Google Scholar - von Rahden BHA, Stein HJ, Puhringer F, Koch I, Langer R, Piontek G, Siewert JR, Hofler H, Sarbia M (2005) Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res 65: 5038–5044
Article CAS PubMed Google Scholar - Wang D, Dubois RN (2004) Cyclooxygenase-2: a potential target in breast cancer. Semin Oncol 31(Suppl 3): 64–73
Article CAS PubMed Google Scholar - Williams CS, Leek RD, Robson AM, Banerji S, Prevo R, Harris AL, Jackson DG (2003) Absence of lymphangiogenesis and intratumoural lymph vessels in human metastatic breast cancer. J Pathol 200: 195–206
Article CAS PubMed Google Scholar - Xu JW, Ikeda K, Yamori Y (2004) Upregulation of endothelial nitric oxide synthase by cyanidin-3-glucoside, a typical anthocyanin pigment. Hypertension 44: 217–222
Article CAS PubMed Google Scholar - Zhang DH, Salto-Tellez M, Chiu LL, Shen L, Koay ESC (2003) Tissue microarray study for classification of breast tumors. Life Sci 73: 3189–3199
Article CAS PubMed Google Scholar
Acknowledgements
This study was supported by grants from the Canadian Breast Cancer Research Alliance and Canadian Breast Cancer Foundation, Ontario Chapter (to PK Lala) and a Translational Breast Cancer Postdoctoral Fellowship from the London Regional Cancer Program (to AV Timoshenko).
Author information
Authors and Affiliations
- Department of Anatomy and Cell Biology, The University of Western Ontario, London, N6A5C1, Ontario, Canada
A V Timoshenko & P K Lala - Department of Biology, The University of Western Ontario, London, N6A5B7, Ontario, Canada
A V Timoshenko - Department of Pathology, The University of Western Ontario, London, N6A5C1, Ontario, Canada
C Chakraborty - Department of Physiology and Pharmacology, The University of Western Ontario, London, N6A5C1, Ontario, Canada
G F Wagner
Authors
- A V Timoshenko
You can also search for this author inPubMed Google Scholar - C Chakraborty
You can also search for this author inPubMed Google Scholar - G F Wagner
You can also search for this author inPubMed Google Scholar - P K Lala
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toP K Lala.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Timoshenko, A., Chakraborty, C., Wagner, G. et al. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer.Br J Cancer 94, 1154–1163 (2006). https://doi.org/10.1038/sj.bjc.6603067
- Revised: 23 February 2006
- Accepted: 28 February 2006
- Published: 28 March 2006
- Issue Date: 24 April 2006
- DOI: https://doi.org/10.1038/sj.bjc.6603067