Subcellular localization and CARD-dependent oligomerization of the death adaptor RAIDD (original) (raw)
Introduction
The execution phase of apoptosis involves the activation of a family of cysteine proteases termed caspases.1,2,3 The fully mature, catalytically active caspases are comprised of a heterotetramer made of two subunits derived from the proteolytic processing of the caspase precursors. In addition to the region that gives rise to the two subunits of the active enzyme, procaspase molecules contain an amino terminal prodomain of varying length. Caspases have been subdivided into two classes on the basis of the length of their prodomains.4 Class I caspases, including caspase-2, -8, -9 and -10, contain long prodomains and class II caspases, including caspase-3, -6 and -7, lack or contain short prodomains. To some extent, this division also reflects a functional hierarchy within the caspase family, with class I caspases lying upstream of class II caspases in the apoptotic cascade.5 Processing of the class II caspases by activated class I caspases leads to degradation of key cellular proteins which results in cell death. Some class II caspases, such as caspase-3, appear also to be involved in a feedback amplification loop by activating certain class I caspases.6
Three related protein-protein interaction domains have been identified in molecules involved in apoptosis and are known as the death domain (DD), death effector domain (DED) and caspase recruitment domain (CARD).7,8,9 These domains have similar structures each consisting of six or seven antiparallel amphipathic α-helices which mediate homotypic association of the molecules involved in apoptosis.10,11,12,13 DDs, found in death receptors and the adaptor molecules that are recruited to them, mediate physical association of receptors and adaptors following ligand-dependent receptor activation. DEDs and CARDs, found in adaptor molecules and the prodomains of class I caspases, have similar functions to each other, mediating oligomerization and recruitment of class I procaspase molecules to death complex associated adaptor molecules.4
The class I caspase, caspase-2,14,15 is activated in response to a variety of apoptotic stimuli including factor withdrawal, γ-irradiation, treatment with etoposide or tumour necrosis factor α (TNFα), and Fas ligation.16,17,18,19 Caspase-2 dimerization and autoprocessing is dependent on its prodomain20 which contains a CARD.9 Caspase-2 associates via its prodomain with RAIDD, an adaptor molecule identified on the basis of its similarity to the prodomain of caspase-2.21,22 RAIDD contains a CARD and a DD at its amino- and carboxyl-termini, respectively.21,22 The interaction between RAIDD and procaspase-2 is believed to be mediated through their respective CARDs,12,21,22 whereas the DD of RAIDD can associate with the DD of RIP, a serine/threonine kinase that interacts with the tumour necrosis factor receptor 1 (TNFR-1) via association with TRADD.21,22,23 Caspase-2 has been shown to associate with the TNFR-1 complex via association with RAIDD when each component of the complex is expressed in 293T cells.22 These data suggest that RAIDD is functionally similar to FADD which recruits caspase-8 to the CD95 death receptor.24,25 The amino terminus of FADD contains a DED which associates with the DEDs present in the prodomain of caspase-8, while the carboxyl terminus contains a DD which associates with the DD of CD95/Fas/Apo-1.24,25,26,27 Upon overexpression, FADD induces apoptosis in transfected cells,26 and forms filaments which have been termed death effector filaments (DEFs).28,29 It has been suggested that DEFs, which can recruit caspase-8, are required for apoptosis induced by DED containing molecules.29
We and others have previously shown that procaspase-2 is localized to both the cytoplasm and nucleus and can be processed in both compartments.5,30 Although the role of caspase-2 in the nucleus is not clear, nuclear caspase-2 is rapidly activated in cells induced to undergo apoptosis.30 Nuclear localization of procaspase-2 is dependent on the prodomain of the molecule,5 however it is not known how nuclear procaspase-2 is activated. Since RAIDD is suggested to be required for the recruitment and activation of caspase-2, it was of interest to determine whether RAIDD colocalizes with caspase-2 in the cell. Additionally, the structural and functional similarities between the DED and CARD led us to compare the ability of these domains to mediate higher order structure formation. Using RAIDD as an example of a CARD-containing adaptor, we have investigated the ability of RAIDD to form higher order structures with similarity to the filamentous structures formed by the DEDs of FADD and caspase-8 in transfected cells.29 In this paper we report the results of such studies.
Results
Subcellular localization of endogenous RAIDD in HeLa and 293T cells
Using two different cell lines and two antibodies, by immunostaining we found that RAIDD was localized throughout the cell. In HeLa cells, a substantial portion of endogenous RAIDD appeared as nuclear, while in 293T cells, most of the RAIDD was cytoplasmic (Figure 1). We further analyzed the distribution of RAIDD in nuclear and cytoplasmic fractions from HeLa and 293T cells. As shown in Figure 2A, from HeLa cells the majority of RAIDD was recovered in the cytoplasmic fraction, whereas a small fraction (∼10%) was associated with the nuclear fractions. In 293T cells, no significant RAIDD was detected in the nuclear fraction (Figure 2B). Since RAIDD has been proposed to recruit caspase-2 to TNFR1, we analyzed the distribution of RAIDD in HeLa cells treated with TNF. Both, immunoblotting of subcellular fractions (Figure 2A) and immunostaining of TNF treated cells (data not shown), failed to show any appreciable changes in RAIDD distribution between the cytoplasm and nucleus.
Figure 1
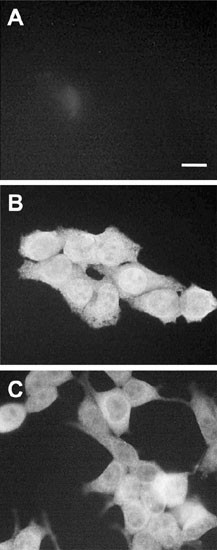
Subcellular localization of RAIDD. HeLa (A, B) or HEK 293T (C) cells were fixed and incubated with an anti-RAIDD polyclonal antibody (Santa Cruz), followed by a FITC-conjugated secondary antibody in (B) and (C). In (A), HeLa cells were incubated with the secondary antibody alone. A similar control with 293T cells (not shown) did not show any background staining. Immunostained cells were visualized by fluorescence microscopy and photographed
Figure 2
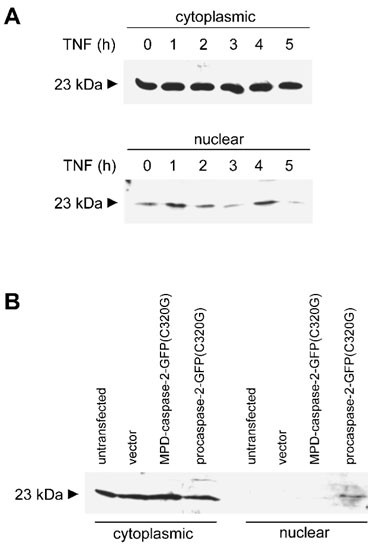
Cytoplasmic and nuclear distribution of RAIDD. (A) The effect of TNFα treatment of HeLa cells on the subcellular distribution of RAIDD. Nuclear and cytoplasmic extracts were prepared from HeLa cells treated with TNFα (10 ng/ml) and cycloheximide (10 μg/ml) for the indicated time, and analyzed by immunoblotting using an anti-RAIDD antibody. Using two different antibodies, RAIDD was detected as a 23 kDa band. Although not shown here, after 4 h treatment with TNFα, >50% of endogenous caspase-2 is fully processed to subunits. (B) Caspase-2 overexpression causes redistribution of RAIDD in HEK 293T cells. Cells were transfected with a GFP expression vector (pEGFP-N1), GFP vector carrying full length catalytically inactive caspase-2 [procaspase-2-GFP(C320G)] or caspase-2(C320G) lacking the prodomain [MPD-caspase-2-GFP(C320G)]. Nuclear and cytoplasmic fractions from untransfected or transfected cells were analyzed by immunoblotting using an anti-RAIDD antibody. Note that unlike in HeLa cells, no significant pool of RAIDD is present in the nucleus of untransfected HEK 293T cells. Immunoblotting with anti-GFP antibody showed significant amounts of expression of caspase-2 fusion proteins (results not shown)
Since HEK 293T cells express relatively lower levels of caspase-2 as compared to HeLa cells (data not shown), we tested whether ectopic expression of procaspase-2 could affect the nuclear/cytoplasmic distribution of RAIDD. We therefore monitored the subcellular localization of RAIDD in 293T cells transfected with a catalytically inactive procaspase-2 expression construct. Indeed, in procaspase-2 transfected 293T cells, the nuclear fraction contained significant levels of RAIDD (Figure 2B, lane 8). When caspase-2 lacking the prodomain was coexpressed with RAIDD, no nuclear RAIDD was evident, suggesting that prodomain-dependent nuclear transport of procaspase-2 was necessary to sequester some RAIDD into the nucleus.
RAIDD colocalizes with caspase-2 in nuclear dots in transfected cells
To further facilitate RAIDD localization and RAIDD/caspase-2 interaction studies, we generated a number of FLAG- or GFP-tagged RAIDD constructs (Figure 3). To ensure that tagging did not affect the activity of RAIDD, the tagged constructs were analyzed in transient cell killing assays. FLAG epitope tagged RAIDD induced significant levels of cell death by 48 h (results not shown) as has been previously seen.21,22 In transfected 293T cells RAIDD-FLAG localized to both the nucleus and cytoplasm showing a diffuse staining pattern (Figure 4A). Similar results were seen in NIH3T3 cells (not shown), suggesting that overexpression of RAIDD may cause some protein to localize in the nucleus. As shown previously,5 procaspase-2-GFP(C320G) localized to the cytoplasm and to discreet dot-like and filamentous structures predominantly in the nucleus of transfected 293T cells (Figure 4B). When cotransfected with procaspase-2-GFP, RAIDD localized to the nucleus and cytoplasm, with some nuclear RAIDD colocalizing with procaspase-2 in dot-like structures (Figure 4C–E). This colocalization was dependent on the prodomain of procaspase-2 since RAIDD and caspase-2 lacking the prodomain (MPD-caspase-2-GFP) did not colocalize in dot-like structures (Figure 4F). These results were confirmed by coimmunoprecipitation analysis. Full length RAIDD could interact with procaspase-2-GFP(C320G) but was unable to interact with MPD-caspase-2-GFP (Figure 4G). These results indicate that RAIDD colocalization with the caspase-2 precursor in the nucleus is dependent on the caspase-2 prodomain.
Figure 3
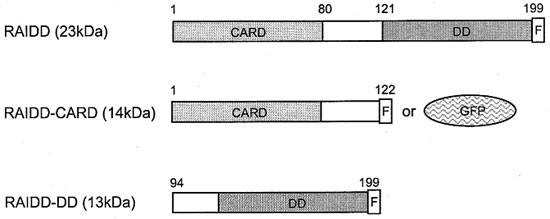
A representation of RAIDD constructs used in transfection experiments. The caspase recruitment domain (CARD), death domain (DD), FLAG epitope (F) and GFP are shown. RAIDD, full length RAIDD with a carboxy terminal FLAG epitope; RAIDD-CARD, truncated RAIDD consisting of the amino terminal 122 amino acids and a carboxy terminal FLAG epitope or GFP fusion; RAIDD-DD, truncated RAIDD consisting of the carboxy terminal 105 amino acids and a carboxyl terminal FLAG epitope
Figure 4
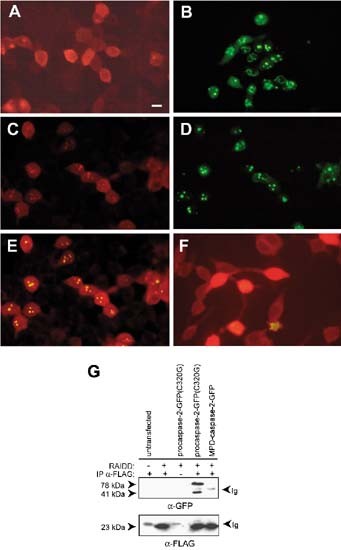
Subcellular localization of RAIDD and procaspase-2 in transfected cells. HEK 293T cells were transiently transfected with (A) RAIDD-FLAG; (B) procaspase-2(C320G)-GFP; (C–E) RAIDD-FLAG and procaspase-2(C320G)-GFP; and (F) RAIDD-FLAG and MPD-caspase-2-GFP. Cells were fixed and immunostained, where appropriate, 18–20 h after transfection and visualized by fluorescence microscopy. RAIDD-FLAG was detected with an anti-FLAG primary antibody and an anti-mouse Ig-rhodamine conjugated secondary antibody. In a group of cells co-transfected with RAIDD-FLAG and procaspase-2(C320G)-GFP the localization of RAIDD-FLAG (C), procaspase-2(C320G)-GFP (D) and RAIDD-FLAG and procaspase-2(C320G)-GFP using a red/green dual filter (E) is shown. In (F) the localization of RAIDD-FLAG and MPD-caspase-2-GFP is shown as in (E). The scale bar represents 15 μm. (G) interactions between RAIDD-FLAG and procaspase-2(C320G)-GFP were detected by immunoprecipitation. HEK 293T cells were transfected with RAIDD-FLAG expression constructs as indicated. Immunoprecipitations were carried out with an unrelated control antibody (lane 3) or anti-FLAG antibody (other lanes). The immunoblot was probed with anti-GFP antibody (upper panel) or anti-FLAG antibody (lower panel). The 78 kDa band corresponds to procaspase-2(C320G)-GFP, the 41 kDa band corresponds to the p14-GFP fragment of caspase-2-GFP, as previously noted,5 and the 23 kDa band corresponds to RAIDD
RAIDD disrupts the formation of caspase-2 prodomain filaments
Upon overexpression, procaspase-2 appears as filamentous and dot-like structures predominantly within the nucleus, whereas the prodomain of caspase-2 appears mostly as an elaborate filamentous network almost exclusively in the nucleus (Figure 5A,B).5 Since the CARD-containing prodomain of caspase-2 is able to dimerise in yeast,20 filament formation is likely to result from CARD-mediated oligomerization of procaspase-2 in transfected cells.5 We were interested to test whether interaction between RAIDD and caspase-2 would affect the caspase-2 prodomain filaments. Indeed, cotransfection of RAIDD with PD-GFP resulted in the disruption of the caspase-2 prodomain filaments (Figure 5C,D). The resulting dot-like structures (Figure 5C,D) contained both PD-GFP and RAIDD (not shown). These results indicated that the association of RAIDD with the prodomain of caspase-2 inhibits prodomain filament formation.
Figure 5
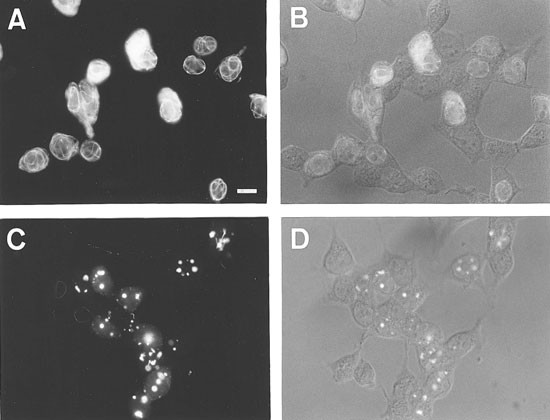
Disruption of PD-GFP filaments by RAIDD in transiently transfected cells. The localization of PD-GFP or PD-GFP and RAIDD-FLAG is shown in HEK 293T cells transfected with (A B) PD-GFP or (C, D) PD-GFP and RAIDD-FLAG (at 1 : 4 ratio). Cells were fixed and visualized by fluorescence microscopy (A, C) or Nomarski optics (B, D) 18 h after transfection. Scale bar represents 15 μm
RAIDD-CARD forms nuclear and cytoplasmic filaments
The recent description of DEFs, which form in cells transfected with DED-containing molecules such as caspase-8 and FADD,28,29 led us to investigate CARD-mediated filament formation. Although the prodomain of caspase-2 localizes in filamentous structures, overexpression of the CARD containing prodomain of caspase-9,5 or DRONC (PA Colussi and S Kumar, 1999, unpublished data) do not, suggesting that not all CARDs have the ability to oligomerize. To investigate whether the RAIDD-CARD was capable of filament formation, RAIDD-CARD and DD were tagged at the carboxy termini with the FLAG epitope or with GFP (Figure 3). In most transfected 293T cells RAIDD-DD was localized diffusely throughout the cytoplasm and in a small proportion of cells RAIDD-DD formed cytoplasmic filamentous structures (Figure 6A). In contrast to full length RAIDD, RAIDD-DD was not seen in the nucleus indicating that nuclear localization of RAIDD is dependent on the CARD-containing region of the protein. Immunoblot analysis of nuclear and cytoplasmic fractions of the transfected cells indicated that the majority of the RAIDD-DD protein was localized in the cytoplasm (Figure 6D). In comparison, the RAIDD-CARD localized to filaments to a large extent within the cytoplasm (Figure 6B,D). The RAIDD-CARD appeared as an extensive filamentous network, sometimes appearing as lariat structures around the nucleus (Figure 6B). RAIDD-CARD filaments were distinguishable from PD-GFP filaments which appear to be predominantly nuclear in localization.5 These data indicate that like the DED, the CARD of RAIDD is able to mediate filament formation. A truncated FLAG epitope tagged RAIDD molecule consisting of the residues 1–80 which lacks helix 6 of the CARD, was unable to form filaments in transfected cells and was localized diffusely throughout the cell (data not shown), suggesting that an intact CARD structure is required for filament formation. From the solution structure of RAIDD-CARD, helix 6 is positioned above the four-helical bundle consisting of helicies 2–5 which forms the core of the CARD12 and may therefore be required to stabilize the CARD structure. Removal of it would therefore be expected to destabilize the CARD structure. We also tested the ability of RAIDD-CARD molecules to interact with each other. The RAIDD-CARD-GFP and RAIDD-CARD-FLAG constructs were cotransfected into 293T cells and proteins immunoprecipitated with an anti-FLAG antibody, followed by immunoblotting with an anti-GFP antibody. In transfected cells colocalization of CARD-GFP and CARD-FLAG was commonly seen (Figure 6E), and the two proteins were coprecipitated (Figure 6F), indicating an interaction between RAIDD-CARD molecules.
Figure 6
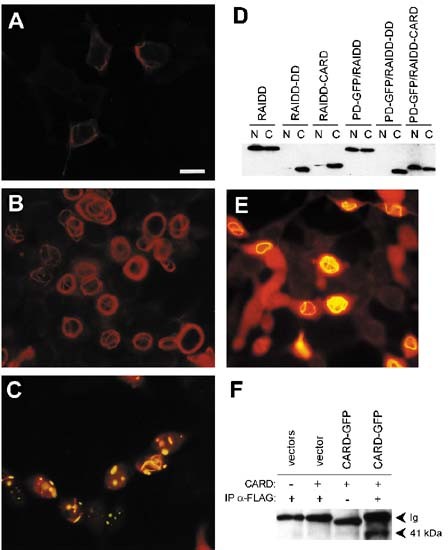
Subcellular localization of RAIDD-CARD and RAIDD-DD in transiently transfected cells. HEK 293T cells were transfected with (A) RAIDD-DD-FLAG; (B) RAIDD-CARD-FLAG; (C) RAIDD-CARD-FLAG and PD-GFP; (E) RAIDD-CARD-GFP and RAIDD-CARD-FLAG. At 18 h post-transfection, cells were fixed, and immunostained prior to visualization. Cells were observed by fluorescence microscopy using a red (A,B) or red/green dual filter (C,E). RAIDD-FLAG proteins were detected with an anti-FLAG primary antibody and anti-mouse Ig-rhodamine conjugated secondary antibody. In (C) and (E) the colocalized proteins appear as orange/yellow dots or filaments. Scale bar represents 15 μm. In (D) nuclear (N) and cytoplasmic (C) extracts from 293T cells transfected as indicated were analyzed by immunoblotting using the anti-FLAG antibody. In (F) extracts from 293T cells transfected as described were immunoprecipitated using an anti-FLAG antibody or an unrelated antibody (lane 3), followed by immunoblotting using an anti-GFP antibody. In lane 1 cells were transfected with vectors pCXN2 and PEGFP-N1; in lane 2, with RAIDD-CARD-FLAG and pEGFP-N1; and in lanes 3 and 4, with RAIDD-CARD-FLAG and RAIDD-CARD-GFP
Caspase-2 prodomain facilitates RAIDD-CARD localization to the nucleus
As shown in Figure 2, procaspase-2 can sequester some endogenous RAIDD to the nucleus. This led us to investigate the localizations of PD-GFP and RAIDD proteins in cotransfected cells. Cotransfection experiments using an excess of PD-GFP and various RAIDD constructs were carried out. Immunoblot analysis of equivalent amounts of nuclear and cytoplasmic extracts from co-transfected cells indicated that the prodomain of caspase-2 does not alter significantly the nuclear/cytoplasmic distribution of either full length RAIDD or RAIDD-DD (Figure 6D). However, consistent with the observations in Figure 2, the cellular distribution of RAIDD-CARD was altered in the presence of the caspase-2 prodomain, with more RAIDD-CARD localizing to the nucleus in cotransfected cells (Figure 6D). When transfected alone, both PD-GFP and RAIDD-CARD form filaments, but in cotransfected cells a strikingly altered localization was seen. In these cells, PD-GFP filaments were disrupted and PD-GFP and RAIDD-CARD colocalized commonly in dot-like structures particularly in the nucleus (Figure 6C). In cells cotransfected with RAIDD-DD and PD-GFP, PD-GFP filament formation was not disrupted and RAIDD-DD was localized diffusely throughout the cytoplasm (data not shown).
Physical interaction between RAIDD-CARD and RAIDD-DD
The CARD and DD of RAIDD are separated by an approximately 20 amino acid linker region. The observation by Chou et al12 that trypsin digestion of full length RAIDD does not result in separation of the protein into two domains led them to suggest that the linker region does not exist as a flexible loop and that the RAIDD molecule exists as a compact unit. The fact that full length RAIDD does not form filaments while the CARD domain can, suggested that the CARD of RAIDD is normally in a conformation that prevents CARD-mediated oligomerization. We investigated the possible interaction between RAIDD-CARD and RAIDD-DD by immunoprecipitating the two domains from 293T cells transfected with RAIDD-CARD-GFP and RAIDD-DD-FLAG. A 41 kD band corresponding to RAIDD-CARD-GFP was detected in cotransfected cells when immunoprecipitation was carried out with the anti-FLAG antibody and immunoblotting was performed with an anti-GFP antibody (Figure 7). These data indicate that the CARD and DD of RAIDD can associate with each other, either directly or indirectly. The ability of the RAIDD-CARD and DD to interact with each other even when expressed as separate proteins provides further evidence that full length RAIDD exists as a compact unit in which the CARD and DD associate.
Figure 7
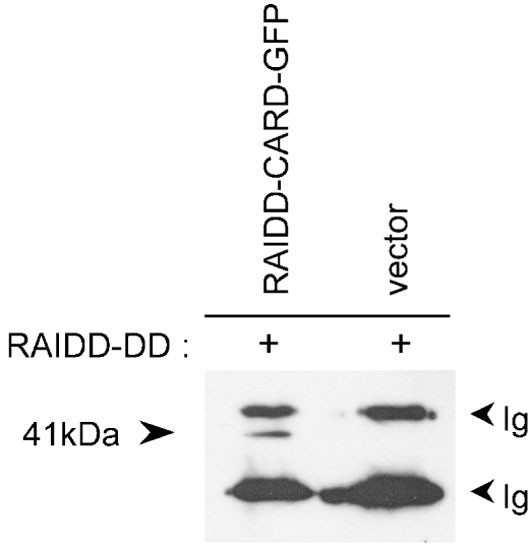
Coimmunoprecipitation of RAIDD-CARD and RAIDD-DD. Cells were transfected with RAIDD-DD-FLAG and RAIDD-CARD-GFP or a control GFP expression vector (pEGFP-N1). Immunoprecipitations were carried out from cotransfected HEK 293T cells with anti-FLAG antibody and the immunoblot probed with anti-GFP antibody
CARD filaments have distinct properties to DED death effector filaments
DEFs observed in cells transfected with DED containing molecules, FADD and caspase-8, do not localize with any known cytoskeletal components suggesting that the filaments may colocalize with an as yet unidentified cytoskeletal component or may form independently due to oligomerization.29 RAIDD-CARD and FADD filaments are similar in appearance and cellular localization. There are conflicting data on the effect of Bcl-xL expression on FADD filaments. While Siegel et al29 found Bcl-xL does not affect formation of FADD filaments, Perez and White28 reported that Bcl-xL effectively blocks generation of these higher order structures. In our experiments, we observed that RAIDD-CARD filament formation was unaffected by Bcl-xL (Figure 8A,B), whereas, Bcl-xL disrupted the formation of FADD filaments (Figure 8C,D). To investigate the possible association of DED and CARD filaments with each other, we coexpressed RAIDD-CARD-GFP with FADD. In a small number of cotransfected cells, RAIDD-CARD and FADD filaments partially colocalized (Figure 8E). Since RAIDD and FADD do not interact directly,22 these results suggest that some higher order structures formed by CARDs and DEDs probably associate through additional proteins.
Figure 8
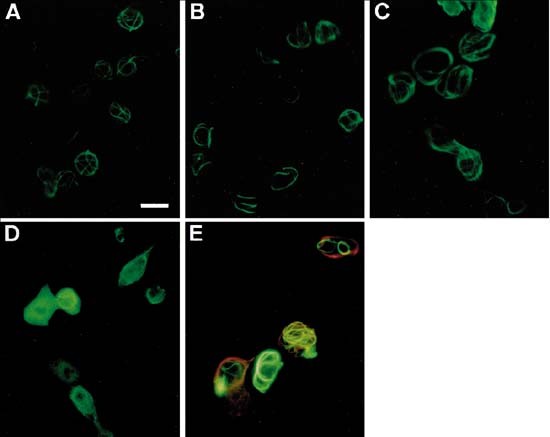
RAIDD-CARD and FADD filaments in transiently transfected cells. The effect of Bcl-xL expression on RAIDD-CARD and FADD filaments (A–D). HEK 293T cells were transfected with RAIDD-CARD-FLAG (A), FADD (C), RAIDD-CARD-FLAG and Bcl-xL (B) or FADD and Bcl-xL (1 : 4) (D). Cells were fixed, immunostained 18 h after transfection and visualized by fluorescence microscopy. RAIDD-CARD-FLAG and FADD were detected with anti-FLAG or anti-FADD primary antibodies respectively and an anti-mouse Ig-FITC conjugated secondary antibody. Colocalization of RAIDD-CARD and FADD filaments in cotransfected cells (E). Cells were cotransfected with equal amounts of RAIDD-CARD-GFP and FADD expression constructs, immunostained 18 h after transfection and visualized by fluorescence microscopy. FADD was detected with an anti-FADD antibody and anti-mouse Ig-rhodamine conjugated secondary antibody. Note that in some transfected cells RAIDD-CARD filaments colocalize with the FADD filaments which appear as orange/yellow structures under the red/green dual filter (E). Scale bar in (A) (representing A–E) equals 15 μm
Discussion
Several recent studies suggest that activation of class I caspases results from oligomerization, often assisted by adaptor molecules that recruit class I caspases to specific death complexes.4,20,31,32,33,34,35,36,37 Oligomerization seems to be mediated by specific domains present in class I caspases and adaptor molecules. It can thus be envisaged that death signals lead to the formation of multiprotein, higher order complexes which primarily function to recruit class I caspases. Consistent with this model, using GFP- and epitope tagged proteins, it has been shown that the prodomain of caspase-8 which contains two DEDs, and either full length FADD or the DED of FADD, form higher order filamentous structures, termed DEFs, in transfected HeLa and 293T cells.28,29 This aggregation seems sufficient to mediate apoptosis in cells overexpressing DEDs from either FADD or caspase-8, by directly recruiting procaspase-8.28,29 In this paper we have shown that similar to the DEDs of FADD and caspase-8, the RAIDD-CARD is also able to form filamentous structures upon overexpression in transfected cells. The physiological significance of these observations is not clear at present as we have not seen formation of higher order structures with endogenous RAIDD. However, endogenous RAIDD filaments may not be observable under normal conditions due to the fact that the DD containing carboxyl terminal half of RAIDD may be involved in inhibiting CARD-mediated aggregation of the RAIDD molecule. This hypothesis is supported by our observation that RAIDD-CARD and RAIDD DD proteins can be coimmunoprecipitated from cells. Thus, it is likely that steric hindrance caused by folding of the RAIDD molecule, due to direct or indirect interaction between amino terminal CARD and carboxyl terminal DD of RAIDD is a mechanism that regulates inappropriate oligomerization of RAIDD. Death signals that promote recruitment of RAIDD through its DD may cause a conformational change which allows CARD-CARD mediated aggregation to occur and thus the formation of higher order structures.
When expressed alone, RAIDD-CARD and the caspase-2 prodomain form elaborate filamentous structures. Interestingly, coexpression of RAIDD-CARD and caspase-2 prodomain caused disruption of filaments, often resulting in colocalization of the two molecules as discrete dots particularly in the nucleus and an increase in nuclear RAIDD-CARD. This result suggests that both RAIDD and caspase-2 filamentous structures result due to oligomerization that can be disrupted by intermolecular interaction between the two molecules. It is noteworthy that unlike the DEDs of FADD and caspase-8,29 the CARDs of RAIDD and caspase-2 do not induce apoptosis upon overexpression. Although RAIDD-CARD filaments have a similar appearance to FADD-DED filaments, they have distinct properties. While Bcl-xL expression disrupts FADD-DED filaments,28 it had no effect on RAIDD filaments. Furthermore, unlike FADD, caspase-8 and caspase-2, overexpression of full length RAIDD does not result in the formation of higher order structures. These results imply that not only the DED and CARD filaments are distinct, the higher order structures formed by different CARD containing molecules also have different properties.
Coexpression of RAIDD and procaspase-2 results in a striking redistribution of RAIDD within the nucleus. Since deletion of the prodomain of caspase-2 abolishes nuclear colocalization, it is reasonable to assume that the interaction via CARDs is necessary for nuclear localization of RAIDD. Although, RAIDD contains a stretch of basic amino acid residues 83REKLKKAR), resembling a nuclear localization sequence38 within the CARD, mutations in this region did not alter the distribution of RAIDD between the nucleus and cytoplasm (data not shown) suggesting that this sequence is insufficient for the nuclear transport of RAIDD. Since more RAIDD is localized in the nucleus in cells expressing high levels of procaspase-2, it is possible that RAIDD is transported to nucleus in a complex with procaspase-2.
It is interesting to note that a number of recently discovered proteins implicated in cell death are localized in the nuclear compartment. DEDD, a DED containing protein, is translocated into nucleus upon stimulation of CD95, where it colocalizes in the nucleolus with UBF, a factor required for RNA polymerase I transcription.39 A DD containing death protein, MyD88, has been shown to localize as fibrillar aggregates, both in the cytoplasm and the nucleus.40 Upon treatment of HeLa cells with TNF, procaspase-1 localizes to the nucleus in a prodomain dependent manner.41 Recently, caspase-9 has also been shown to translocate from the mitochondria to nucleus upon induction of apoptosis.42 Apoptosis inducing factor (AIF), a flavoprotein that is released from mitochondria during apoptosis, functions by mediating changes in the nucleus43 and is thus likely to be imported into nucleus. These findings suggest that a number of proteins, including caspase-2 and RAIDD, may participate in nuclear apoptotic functions not previously envisaged.
From overexpression studies, RAIDD has been proposed to recruit procaspase-2 to TNFR1 and CD95 via association with RIP and TRADD.21,22 However, several lines of evidence suggest that caspase-2 and RAIDD do not play a major role in either TNF or CD95L mediated apoptotic pathways. For instance, caspase-2 mutant mice do not exhibit a defect in TNF or CD95L mediated cell death.44 Additionally, cells from mice carrying a targeted disruption of RIP, the molecule that has been proposed to recruit RAIDD to TNFR-1 via the adaptor TRADD,21,22 are not resistant to TNF-induced apoptosis.45 Instead, RIP deficient mice are defective in NF-κB activation, suggesting that RIP is required for the TNF-mediated cell survival pathway, not the death pathway.45 Finally, disruption of the caspase-8 gene in mice completely abolishes CD95L and TNF induced death, indicating that caspase-8 is the essential class I caspase involved in these pathways.46 The observation that RAIDD and caspase-2 can both localize to the nucleus, in addition to the cytoplasmic compartment, raises the possibility that the molecular interactions between these two proteins may be necessary for as yet unidentified nuclear and cytoplasmic function(s). However, from our data and those reported earlier21,22 it is not entirely clear whether RAIDD is necessary for caspase-2 activation during apoptosis. Based on these observations we propose that the interaction between RAIDD and caspase-2 is involved in a process such as recruitment to specific subcellular structures, rather than caspase activation.
Many recent studies have emphasized the importance of oligomerization through specific protein-protein interaction domains in apoptotic signalling.4 The data presented in this paper further support this concept. Since some of these studies are based on overexpression of proteins in transfected cells, one can argue that CARD-mediated formation of elaborate higher order structures may not occur under physiological conditions where the concentration of CARD containing proteins such as RAIDD and procaspase-2 are unlikely to be high. While it is conceivable that extensive aggregates of CARD- and DED-containing proteins may not form under physiological situations (as also evident from endogenous RAIDD localization studies presented in this study), based on the fact that these domains are able to oligomerize very efficiently in vivo, it is possible that localized high concentrations of these proteins generate transient higher order structures in response to death signalling. Although we have not yet seen formation of higher order RAIDD structures in response to apoptotic stimuli, a recent study has found that endogenous FADD and caspase-8 form filamentous structures in cycloheximide treated Jurkat cells.47
Materials and Methods
Expression vectors
The coding region of human RAIDD was amplified by PCR from reverse transcribed Jurkat cell mRNA and cloned into pBluescript to generate pBSRAIDD. Wild type and mutant RAIDD cDNAs were amplified from pBSRAIDD by polymerase chain reaction in the following manner. Full length RAIDD was amplified with primer A (5′-CGGAATTCATGGAGGCCAGAGACAAA) and primer B (5′-CGGGAATTCTCACTTGTCATCGTCGTCCTTGTAGTCCTCCAACATGTCATGTGCAGGAG) which contains the coding sequence for FLAG epitope (sequence homologous to RAIDD is underlined). Primers C (5′-CGGGAATTCGCCATGACCGACCTGCCTGC) and D (5′-CGGGAATTCTCACTTGTCATCGTCGTCCTTGTAGTCCTGGTTAATCTAATCTGCCGGTC) were used for amplification of carboxyl- (RAIDD-DD), amino-terminal (RAIDD-CARD) regions of RAIDD, respectively. Amplified products were digested with _Eco_RI and cloned into the _Eco_RI digested mammalian expression vector pCXN2. The RAIDD-ΔCARD construct was amplified using primers A and E (5′-CGGGAATTCTCACTTGTCATCGTCGTCCTTGTAGTCGGGAAACGAAACTCCTGTAGG), digested with _Eco_RI and cloned into _Eco_RI digested pCXN2. Point mutations were introduced into the sequence of RAIDD using the Quikchange (Stratagene) method. The RAIDD-CARD-GFP construct was amplified using primers A and F (5′-CCGGTGGTACCTGGTTAATCTGCCG), digested with _Eco_RI and _Kpn_I and cloned into pEGFP-N1 (CLONTECH) digested with the same enzymes. Caspase-2-GFP(C320G), PD-GFP and MPD-GFP have been described previously.5 The FADD and Bcl-xL expression vectors were kindly provided by Drs. A Strasser and D Huang.
Cell culture and transfection
NIH3T3 and HeLa cells were grown in DMEM containing 10% foetal calf serum. HEK 293T cells were maintained in Dulbecco's modified Eagle's medium and 1640 RPMI supplemented with 10% foetal calf serum. For immunoblotting cells were plated at a density of 5×105 NIH3T3 or HeLa cells or 1×106 293T cells/60 mm dish, grown for 24 h and transfected with 2 μg of plasmid DNA, where appropriate, using the FuGENE 6 reagent according to manufacturer's (Boehringer Mannheim) protocol. For coimmunoprecipitation, HEK 293T cells were plated at a density of 2.5×106 cells/100 mm dish, grown for 24 h and transfected with a total of 4 μg DNA (equal amounts of two expression vectors) using the FuGENE 6 transfection reagent (Boehringer Mannheim). Cells were harvested 20–24 h post-transfection.
Immunohistochemical techniques and microscopy
For microscopy, cells were fixed in 4% paraformaldehyde (PFA) for 30 min. For immunohistochemistry, 293T and HeLa cells were fixed in 4% PFA, followed by 15 min in 47.5% methanol, 47.5% acetone and 5% formaldehyde, whereas NIH3T3 cells were fixed for 30 min in 47.5% methanol, 47.5% acetone and 5% formaldehyde. Antibodies were diluted in 1% foetal calf serum in PBS. The two RAIDD polyclonal antibodies were purchased from SantaCruz and StressGen respectively. The anti-FLAG M2 monoclonal antibody was from Sigma, anti-FADD from Transduction Laboratories, anti-GFP monoclonal antibody, anti-rabbit Ig-FITC, anti-mouse Ig-FITC and anti-mouse Ig-rhodamine from Boehringer Mannheim. Cells were viewed and photographed using a fluorescence microscope (Olympus BH2-RFCA).
Immunoblotting and immunoprecipitation
Cellular subfractionation was carried out as previously described.5 For immunoprecipitation, cells were lysed on ice in 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% NP-40 and CompleteTM protease inhibitors (Boehringer Mannheim). Cell extracts were precleared with protein G sepharose (Pharmacia) and antibody binding was carried out overnight at 4°C. Immunoprecipitation was carried out with protein G sepharose using an anti-FLAG M2 monoclonal antibody (Sigma), or a control mouse Ig (kindly provided by Dr. Leonie Ashman). Equivalent amounts of cytosolic and nuclear extract or immunoprecipitation reactions were resolved on 12 or 15% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (DuPont). Membranes were probed with an anti-RAIDD polyclonal, anti-GFP monoclonal or the anti-FLAG M2 monoclonal antibody followed by an appropriate horseradish peroxidase-coupled secondary antibody (Amersham Pharmacia Biotech). Signals were detected using the ECL system (Amersham Pharmacia Biotech).
Abbreviations
CARD:
caspase recruitment domain
DD:
death domain
DED:
death effector domain
DEF:
death effector filaments
GFP:
Aequorea victoria green fluorescent protein
procaspase-2:
the 51-kDa precursor of mouse Nedd2
PD:
prodomain of caspase-2
MPD:
caspase-2 lacking the prodomain region
References
- Thornberry NA and Lazebnik Y . (1998) Caspases: enemies within. Science 281: 312–316
Article Google Scholar - Cryns V and Yuan J . (1998) Proteases to die for. Genes Dev. 12: 1551–1570
Article CAS Google Scholar - Kumar S . (1999) Regulation of caspase activation in apoptosis: Implications in pathogenesis and treatment of disease. Clin. Expt. Pharmacol. Physiol. 26: 295–303
Article CAS Google Scholar - Kumar S and Colussi PA . (1999) Prodomains-adaptors-oligomerization: the pursuit of caspase activation in apoptosis. Trends Biochem. Sci. 24: 1–4
Article CAS Google Scholar - Colussi PA, Harvey NL and Kumar S . (1998) Prodomain-dependent nuclear localization of the caspase-2(Nedd2) precursor. A novel function for a caspase prodomain. J. Biol. Chem. 273: 24535–24542
Article CAS Google Scholar - Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang H-G, Reed JC, Nicholson DW, Alnemri ES, Green DR and Martin SJ . (1999) Ordering of cytochrome c-initiated caspase cascade: Hierarchical activation of caspase-2, -3, -6, -7, -8 and -10 in a caspase-9-dependent manner. J. Cell Biol. 144: 281–292
Article CAS Google Scholar - Ashkenazi A and Dixit VM . (1998) Death receptors: signaling and modulation. Science 281: 305–308
Article Google Scholar - Nagata S . (1997) Apoptosis by death factor. Cell 88: 355–365
Article CAS Google Scholar - Hofmann K, Bucher P and Tschopp J . (1997) The CARD domain: a new apoptotic signalling motif. Trends Biochem. Sci. 22: 155–156
Article CAS Google Scholar - Huang B, Eberstadt M, Olejniczak ET, Meadows RP and Fesik SW . (1996) NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 384: 638–641
Article CAS Google Scholar - Eberstadt M, Huang B, Chen Z, Meadows RP, Ng S-I, Zheng L, Lenardo MJ and Fesik SW . (1998) NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 392: 941–945
Article CAS Google Scholar - Chou JJ, Matsuo H, Duan H and Wagner G . (1998) Solution structure of the RAIDD-CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 94: 171–180
Article CAS Google Scholar - Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES and Shi Y . (1999) Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399: 549–557
Article CAS Google Scholar - Kumar S, Kinoshita M, Noda M, Copeland NG and Jenkins NA . (1994) Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1β-converting enzyme. Genes Dev. 8: 1613–1626
Article CAS Google Scholar - Wang L, Miura M, Bergeron L, Zhu H and Yuan J . (1994) Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 78: 739–750
Article CAS Google Scholar - Harvey NL, Butt AJ and Kumar S . (1997) Functional activation of Nedd2/ICH-1 (caspase-2) is an early process in apoptosis. J. Biol. Chem. 272: 13134–13139
Article CAS Google Scholar - MacFarlane M, Cain K, Sun X-M, Alnemri ES and Cohen GM . (1997) Processing/activation of at least four interleukin-1β-converting enzyme-like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J. Cell. Biol. 137: 469–479
Article CAS Google Scholar - Li H, Bergeron L, Cryns V, Pasternack MS, Zhu H, Shi L, Greenberg A and Yuan J . (1997) Activation of caspase-2 in apoptosis. J. Biol. Chem. 272: 21010–21017
Article CAS Google Scholar - Harvey KF, Harvey NL, Michael JM, Parasivam G, Waterhouse N, Alnemri ES, Watters D and Kumar S . (1998) Caspase-mediated cleavage of the ubiquitin-protein ligase Nedd4 during apoptosis. J. Biol. Chem. 273: 13524–13530
Article CAS Google Scholar - Butt AJ, Harvey NL, Parasivam G and Kumar S . (1998) Dimerization and autoprocessing of the Nedd2(caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J. Biol. Chem. 273: 6763–6768
Article CAS Google Scholar - Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwak G, Fernandes-Alnemri T and Alnemri ES . (1997) CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res. 57: 615–619
CAS PubMed Google Scholar - Duan H and Dixit VM . (1997) RAIDD is a new ‘death’ adaptor molecule. Nature 385: 86–89
Article CAS Google Scholar - Hsu H, Xiong J and Goeddel DV . (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa β activation. Cell 81: 495–504
Article CAS Google Scholar - Boldin MP, Goncharov TM, Goltsev YV and Wallach D . (1996) Involvement of MACH, a novel Mort1/FADD-interacting protease, in Fas/APO1- and TNF receptor-induced cell death. Cell 85: 803–815
Article CAS Google Scholar - Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Scaffidi C, Bretz JD, Zhang M, Ni J, Gentz R, Mann N, Krammer PH, Peter ME and Dixit VM . (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 85: 817–827
Article CAS Google Scholar - Chinnaiyan AM, O'Rourke K, Tewari M and Dixit VM . (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505–512
Article CAS Google Scholar - Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH and Peter ME . (1997) FLICE is activated by association with the CD95 death-inducing signalling complex (DISC). EMBO J. 16: 2794–2804
Article CAS Google Scholar - Perez D and White E . (1998) E1B 19K inhibits Fas-mediated apoptosis through FADD-dependent sequestration of FLICE. J. Cell Biol. 141: 1255–1266
Article CAS Google Scholar - Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J and Lenardo MJ . (1998) Death-effector filaments: Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 141: 1243–1253
Article CAS Google Scholar - Zhivotovsky B, Samali A, Gahm A and Orrenius S . (1999) Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 6: 644–651
Article CAS Google Scholar - Muzio M, Stockwell BR, Stennicke HR, Salvesen GS and Dixit VM . (1998) An induced proximity model for caspase-8 activation. J. Biol. Chem. 273: 2926–2930
Article CAS Google Scholar - Martin DA, Siegel RM, Zheng L and Lenardo MJ . (1998) Membrane oligomerization and release activates the caspase 8 (FLICE/MACHα1) death signal. J. Biol. Chem. 273: 4345–4349
Article CAS Google Scholar - MacCorkle RA, Freeman KW and Spencer DM . (1998) Synthetic activation of caspases: artificial death switches. Proc. Natl. Acad. Sci. U.S.A. 95: 3655–3660
Article CAS Google Scholar - Srinivasula SM, Ahmed M, Fernandes-Alnemri T and Alnemri ES . (1998) Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1: 949–957
Article CAS Google Scholar - Yang X, Chang HY and Baltimore D . (1998) Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell. 1: 319–325
Article CAS Google Scholar - Yang X, Chang HY and Baltimore D . (1998) Essential role of CED-4 oligomerisation in CED-3 activation and apoptosis. Science 281: 1355–1357
Article CAS Google Scholar - Colussi PA, Harvey NL, Shearwin-Whyatt LM and Kumar S . (1998) Conversion of procaspase-3 to an autoactivating caspase by fusion to the caspase-2 prodomain. J. Biol. Chem. 273: 26566–26570
Article CAS Google Scholar - Nigg EA . (1997) Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature 386: 779–786
Article CAS Google Scholar - Stegh AH, Schickling O, Ehret A, Scaffidi C, Peterhansel C, Hofmann TG, Grummt I, Krammer PH and Peter ME . (1998) DEDD, a novel death effector domain-containing protein targeted to the nucleolus. EMBO J. 17: 5974–5986
Article CAS Google Scholar - Jaunin F, Burns K, Tschopp J, Martin TE and Fakan S . (1998) Ultrastructural distribution of the death-domain-containing MyD88 protein in HeLa Cells. Exp. Cell Res. 243: 67–75
Article CAS Google Scholar - Mao P-L, Jiang Y, Wee BY and Porter AG . (1998) Activation of caspase-1 in the nucleus requires nuclear translocation of pro-caspase-1 mediated by its prodomain. J. Biol. Chem. 273: 23621–23624
Article CAS Google Scholar - Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G and Reed JC . (1999) Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 96: 5752–5757
Article CAS Google Scholar - Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM and Kroemer G . (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446
Article CAS Google Scholar - Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws A, Salter JCM, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL and Yuan J . (1998) Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 12: 1304–1314
Article CAS Google Scholar - Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ and Leder P . (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8: 297–303
Article CAS Google Scholar - Varfolomeev EE, Schuchmann M, Luria V, Chiannikulchai N, Beckmann JS, Mett L, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Tama S, Avraham KB, Goncharov T, Holtmann H, Lonai P and Wallach D . (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276
Article CAS Google Scholar - Tang D, Lahti JM, Grenet J and Kidd VJ . (1999) Cycloheximide-induced T-cell death is mediated by a Fas-associated death domain-dependent mechanism. J. Biol. Chem. 274: 7245–7252
Article CAS Google Scholar
Acknowledgements
We thank Andreas Strasser and David Huang for the provision of material, and Paul Colussi for helpful discussions. This work was supported by funds from the National Health and Medical Research Council of Australia, the Wellcome Trust and the Anti-Cancer Foundation of South Australia. LM Shearwin-Whyatt is a Peter Nelson Leukaemia Research Associate. NL Harvey is an Anti-Cancer Foundation Research Associate. S Kumar is a Wellcome Trust Senior Fellow in Medical Science.
Author information
Authors and Affiliations
- Hanson Centre for Cancer Research, Institute of Medical and Veterinary Science, Frome Road, Adelaide, Australia
L M Shearwin-Whyatt, N L Harvey & S Kumar - Department of Medicine, The University of Adelaide, Australia
S Kumar
Authors
- L M Shearwin-Whyatt
You can also search for this author inPubMed Google Scholar - N L Harvey
You can also search for this author inPubMed Google Scholar - S Kumar
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toS Kumar.
Additional information
Edited by S Martin
Rights and permissions
About this article
Cite this article
Shearwin-Whyatt, L., Harvey, N. & Kumar, S. Subcellular localization and CARD-dependent oligomerization of the death adaptor RAIDD.Cell Death Differ 7, 155–165 (2000). https://doi.org/10.1038/sj.cdd.4400632
- Received: 07 July 1999
- Revised: 13 September 1999
- Accepted: 02 November 1999
- Published: 07 March 2000
- Issue Date: 01 February 2000
- DOI: https://doi.org/10.1038/sj.cdd.4400632