The ‘harmless’ release of cytochrome c (original) (raw)
Ever since the early discovery that mitochondria enriched fractions are necessary for induction of apoptosis in cell free systems,1 that Bcl-2 was localized mainly to the outer mitochondrial membrane,2,3 and that mitochondria can lose membrane potential during apoptosis,4 mitochondria have been the focus of apoptosis research. The observation that cytochrome c was involved in activation of caspases5,6 and the demonstration that proapoptotic Bcl-2 family members (e.g. Bid, Bax, Bik and Bok) mediate the translocation of cytochrome c from mitochondria to cytosol,7,8,9,10 an event that is blocked by anti apoptotic Bcl-2 family members (e.g. Bcl-2 and Bcl-xL),11,12,13 firmly established the mitochondrion as a central life and death regulator.
In the cytosol, holocytochrome c initiates the formation of a complex known as ‘the apoptosome’ by binding to an adaptor molecule Apaf-1. This triggers the oligomerization of Apaf-1 followed by recruitment and activation of procaspase-9.14,15 Activated caspase-9 then cleaves and activates the downstream effectors of apoptotic cell death, caspases 3,6 and 7.16 Recently, evidence has emerged that the key components of this pathway have been widely conserved during evolution.14,17,18,19 Therefore, the release of cytochrome c from mitochondria to cytosol can now be regarded as a key regulatory event in apoptosis induced by a wide variety of stimuli. It appears however, that apoptosis induced by ligation of death receptors such as Fas may not require mitochondrial involvement in certain cell types.20
For a while, the release of cytochrome c was considered by many to be a point of no return and it did not matter if mitochondria were damaged in the process of cytochrome c release (since caspase activation would ensure fast and ordered destruction of the dying cell anyway). Recently IAP proteins have been shown to inhibit caspase activation downstream of cytochrome c release,21,22 HSPs have been shown to modulate caspase activation23,24,25 and some cells can recover after cytochrome c release.26,27,28 This hints that cytochrome c release may not always commit a cell to die.
This appears surprising at first glance since cytochrome c and the integrity of the mitochondrial intermembrane space would appear to be essential for respiration and cell survival. Some cells do survive after cytochrome c release and therefore may be without respiration (cell survival in the absence of respiration has been observed in rho° cells 29,30), however in order for mitochondria to exert their metabolic fuctions such as amino acid, heme and steroid metabolism, the membrane potential, protein import function and integrity of the inner membrane have to be maintained after cytochrome c release. It is therefore obvious that for cells to survive the release of cytochrome c, the mechanism of this release must be delicate enough to allow for the recovery of mitochondria provided that downstream events such as caspase activation are blocked.
In this review we will summarize the accumulating evidence that mitochondrial structure and function are well preserved after cytochrome c release in many experimental systems and discuss the mechanistic implications of this finding. We will also discuss the kinetics of cytochrome c release, the possible order of events in apoptotic cells and possible reasons for the loss of mitochondrial function being associated with cytochrome c release in some experimental settings.
Cytochrome c release from isolated mitochondria
Isolated mitochondria provide a model system that has the natural protein and lipid composition of the outer membrane, and therefore reflect the target of cytochrome c releasing factors in the cell. Thus using isolated mitochondria and recombinant proapoptotic Bcl-2 proteins it should be possible to describe events that are associated with cytochrome c release that is mediated by proapototic Bcl-2 family proteins, and to distinguish between events that are sufficient and/or necessary for the release. An advantage of isolated mitochondria is the elimination of secondary events caused by cytosolic factors such as activated caspases.
Mitochondrial membrane potential (Δψm) is produced by the respiratory chain that uses the free energy of substrate oxidation to pump protons out of the matrix. This electrochemical potential across the inner membrane is used by ATP-synthase to produce ATP from ADP and Pi. Since mitochondria form the main ATP producing system of the cell, Δψm is essential for efficient ATP production under aerobic conditions. Δψm is also required for mitochondrial protein import and regulated metabolite transport. Because mitochondria are involved in pathways metabolising steroids, heme, pyrimidines and amino acids, Δψm is often used as an indication of cellular viability.
In one of the first papers to describe cytochrome c release, Newmeyer and colleagues11 used a Xenopus cell free system to show that Δψm was not lost when cytochrome c was released, indicating that mitochondria remain relatively intact. Further work showed that cytochrome c was released from Xenopus mitochondria in a process involving only a limited permeabilization of the outer membrane.31 The limited degree of outer membrane permeabilization in this system was further demonstrated by the observation that a cytosolic activity (named PEF for permeability enhancing factor) can enhance the limited permeability of the outer membrane caused by recombinant Bid or Bax.31 The outer membranes of Bid or Bax treated mitochondria are permeable to cytochrome c, but the rate of cytochrome c traffic across the outer membrane (measured by oxidation of added cytochrome c catalyzed by cytochrome c oxidase) can be further enhanced by PEF. However, even the PEF-enhanced permeabilization of the outer membrane does not lead to damage of the inner membrane.31 Recently von Ahsen et al. demonstrated that recombinant Bid and Bax can release cytochrome c from mitochondria without causing mitochondrial depolarization or inducing changes in mitochondrial ultrastructure.32 Mitochondria that have lost their cytochrome c were shown to be in the same condensed conformation as control mitochondria. Electron microscopy as well as tomography33 could not reveal any significant change in the ultrastructure. No holes, tears or ruptures in the outer membrane could be detected. This suggests a very specific release mechanism possibly involving a very small pore, too small to be visible by electron microscopy. Using mitochondrial protein import as an assay for mitochondrial membrane potential they could also rule out artifacts from other assays such as those observed when some fluorescent dyes were used to measure membrane potential. One problem of the dye uptake assay is the influence of the plasma membrane potential.34 The uptake of positively charged lipophilic dyes like rhodamine-123 has also been questioned because artifacts may arise due to self-quenching.35 Furthermore, rhodamine 123 inhibits complex V,36 while Mito Tracker orange and DioC6(3) inhibit complex I of the respiratory chain.37,38 The posttranslational import of nuclear-encoded proteins into mitochondria however is a vital, natural function of mitochondria. The work of von Ahsen et al.32 demonstrated that protein import was preserved after complete release of cytochrome c. This could be shown for isolated mitochondria from Xenopus oocytes and human HL-60 cells. Mitochondrial membrane potential was also maintained in apoptotic HeLa cells that were induced to undergo apoptosis by treatment with UV or staurosporine, provided that caspase activation was prevented by zVAD-fmk. Furthermore, after permeabilization of the plasma membrane by digitonin, the protein import function of mitochondria was shown to be maintained in apoptotic cells after cytochrome c release.
Others have published data that confirm a subtle mechanism of cytochrome c release in other systems: For example Jurgensmeier et al.9 showed that recombinant Bax could induce cytochrome c release from isolated rat liver mitochondria without swelling, suggesting that a mechanical disruption of the outer membrane is not the mechanism. Similarly, Finucane et al.10 have shown that Bax can induce cytochrome c release from isolated mouse liver mitochondria without swelling. Furthermore it was shown that mitochondrial depolarization in apoptotic 293T cells after Bax expression was a secondary event due to caspase activation since it could be blocked by coexpression of XIAP or addition of zVAD-fmk. This result provides an explanation for mitochondrial depolarization often observed in apoptotic cells.
Cellular studies confirm a non-disruptive mechanism of cytochrome c release
The dynamic interaction of mitochondria with cells has made it difficult to objectively compare cytochrome c release with other events that occur during apoptosis. This has been compounded by the nature of the techniques used to assess cytochrome c release in cells (cellular fractionation followed by Western blotting or immunocytochemistry). Cellular fractionation generally involves mechanical disruption of cells and often results in incomplete rupturing of a large proportion of cells or excessive disruption of many of the mitochondria. Subsequent analysis of the fractions by Western blotting can therefore only be regarded as qualitative. Immunocytochemistry is quantitative; however the fixing process used in this technique limits the mitochondrial parameters other than cytochrome c release that can be assessed. For instance using immunocytochemistry, it is not possible to determine whether mitochondria that have released cytochrome c in cells can maintain protein import and many of the dyes used to measure Δψm are released from the mitochondria upon fixation. In addition, dyes that measure Δψm and can withstand fixation have been shown to induce mitochondrial permeability transition37 and many of these dyes inhibit various aspects of mitochondria metabolism (e.g. Rhodamine 123 inhibits ATP synthase,36 and MitoTracker orange and DioC6(3) inhibit complex I,37,38 as discussed).
More recently, green fluorescent protein (GFP)-tagged cytochrome c expressed in HeLa cells was shown to localize to the mitochondria and to be released concomitantly with endogenous cytochrome c during apoptosis.39 The release of cytochrome _c_-GFP was complete in that it was evenly distributed throughout the cell. This excludes the involvement of an active export mechanism since cytochrome _c_-GFP appeared at the same concentration both inside and outside the mitochondria. Further, the addition of the caspase inhibitor zVAD-fmk did not prevent the release of cytochrome _c_-GFP during cytotoxic drug induced apoptosis, confirming previous data showing that cytochrome c release is caspase independent. These studies clearly demonstrate that mitochondria do not maintain a large proportion of their cytochrome c during apoptosis and exclude the possibility that some mitochondria release cytochrome c via a destructive mechanism (such as membrane rupture) while a subpopulation of mitochondria evade the cytochrome c releasing signal to aid in cell recovery (if required) after cytochrome c release. At least in these cells, all of the mitochondria release most of their cytochrome c.
Using these cells treated with a variety of apoptosis-inducing stimuli, it was shown that mitochondria which have released cytochrome _c_-GFP do not take up TMRE (a fluoresecnt dye used to measure Δψm). However, there were no circumstances where cells dying by apoptosis had a low membrane potential prior to the release of cytochrome _c_-GFP in the mitochondria. Further, in the presence of the caspase inhibitor zVAD-fmk, mitochondria of cells treated with a variety of stimuli maintained Δψm (took up TMRE) for an extended period of time after they had released cytochrome _c_-GFP into the cytoplasm. In the cells that had released cytochrome c in the presence of zVAD-fmk, the Δψm was maintained in the presence of oligomycin (an inhibitor of ATP synthase) but was not maintained in the presence of sodium azide (an inhibitor of complex IV of the electron transport chain), suggesting that even after cytochrome c release Δψm is maintained by the electron transport chain. Since the contribution of complex IV to the Δψm is dependent on acceptance of electrons from cytochrome c, this suggests that even when cytochrome c is diffuse through the cell, the concentration is sufficient to maintain respiration. These data demonstrate that the loss of Δψm is not the cause of cytochrome c release as is predicted in some hypotheses, but is rather a consequence of cytochrome c release that is probably mediated by caspase-proteases. These data also showed clearly that mitochondria can survive the release of cytochrome _c_-GFP without compromising the integrity of the inner mitochondrial membrane. In support of this, cytochrome c release has been reported in HL-60 cells and neuronal cells treated with various death-inducing stimuli without a detectable loss of Δψm40,41 and addition of the caspase inhibitor zVAD-fmk prevented Δψm loss in HeLa cells treated with UV but did not inhibit cytochrome c release.42 These studies in cells also confirmed the oservations made using isolated mitochondria treated with pro-apoptotic Bcl-2 family members (as discussed previously).
Time lapse confocal microscopy using HeLa cells expressing cytochrome _c_-GFP has greatly extended our current knowledge of the mechanism of cytochrome c release in cells. These studies showed irrefutably that cytochrome c release is an early event during apoptosis occurring hours before phosphatidylserine exposure and loss of plasma membrane integrity. These studies also showed that although cytochrome _c_-GFP release is an early event, mitochondria retain cytochrome _c_-GFP for many hours after the initial apoptosis-inducing stimulus, however once initiated all mitochondria release cytochrome _c_-GFP in approximately 5 min regardless of the nature of the initial stimulus. The relatively invariable duration of cytochrome _c_-GFP release (∼5 min) indicates that a universal pathway of cytochrome c release may be used during cytotoxic drug-induced and TNF-induced apoptosis or that the mechanisms of release are remarkably similar.
During TNF induced apoptosis, caspase-8 is activated early and mediates the release of cytochrome c via the activation of the pro-apoptotic Bcl-2 family member Bid.7,8 This has led to the speculation that during apoptosis induced by cytotoxic drugs (that rely on the release of cytochrome c for caspase activation), caspases may signal for increased cytochrome _c_-release. The addition of zVAD-fmk inhibited the release of cytochrome _c_-GFP during TNF-induced apoptosis but did not extend the duration of cytochrome c release during apoptosis induced by other stimuli, thus confirming the participation of caspases in the release of cytochrome c during TNF induced apoptosis.39 However the involvement of caspases in an amplification loop during apoptosis induced by other stimuli is unlikely in these cells.
Goldstein et al.39 also showed that duration of cytochrome _c_-GFP release from mitochondria was similar over a temperature range of 13°C. Since the activity of most enzymes changes dramatically over 10°C, if enzymes were rate limiting during cytochrome c release, lower temperatures would be expected to slow the period over which cytochrome c is released. Individual mitochondria or groups of mitochondria were shown to release cytochrome c within 2 min and therefore the duration of cytochrome c release in cells was reported to reflect the time between cytochrome c release from the first and last mitochondria in the cell. At lower temperatures a slowed enzymatic process would result in cytochrome c release from the last mitochondria with a delayed kinetics after the first (if enzyme kinetics is slowed and plays a critical role in the kinetics), resulting in an apparent extension of the duration of cytochrome c release. Since an increase in duration was not observed, this reasoning suggested that the direct mechanism of cytochrome c release was temperature independent and did not rely on enzymes.
From the experiments performed on isolated mitochondria as well as in living cells, we conclude that proapoptotic Bcl-2 family members are sufficient to release cytochrome c and other proteins from the intermembrane space without causing severe damage seen as depolarization and swelling of mitochondria. This is clearly inconsistent with all models based on matrix swelling and outer membrane rupture (caused by either permeability transition or hyperpolarization) as the primary mechanism of cytochrome c release in apoptotic cells.
The question arises of why mitochondria can keep their membrane potential high after cytochrome c release. If cytochrome c is not actively transported, it will only equilibrate within the total cell volume. Under conditions that require no or only low ATP synthase activity, the residual cytochrome c concentration might be sufficient to sustain respiration. So using isolated mitochondria in buffer, the dilution may be limiting for respiration. In cells, this dilution would be determined by mitochondrial content and cytoplasmic volume of the cell. Another factor may be the rate of ATP consumption in the cell. If ATP is consumed quickly, the remaining membrane potential may be used up, since the respiratory chain will not work efficiently under conditions of cytochrome c depletion. Some cells however may even compensate for low or absent respiration by enhanced glycolysis and reverse action of complex V to maintain mitochondrial Δψm. Thus if glucose is sufficient, cells can maintain Δψm and thus may be able to survive cytochrome c release.
Possible mechanisms of cytochrome c release
Data from our lab and from others clearly show that cytochrome c release can happen in a very subtle way leaving much of mitochondrial structure and function intact. What kind of a mechanism may account for such a gentle process? Some mechanisms for cytochrome c release have been proposed, among them very disruptive ways but also some ways of limited outer membrane permeabilization (Figure 1)
Figure 1
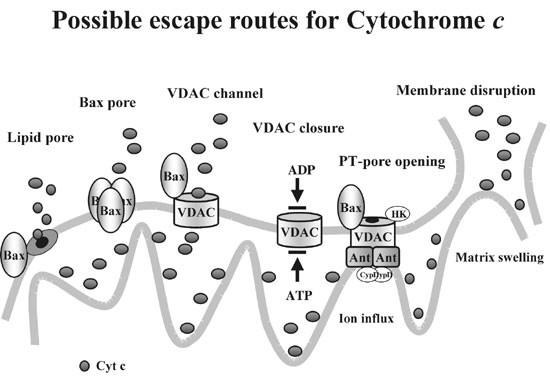
Models of cytochrome c release. Two of these models rely on matrix swelling causing mechanical disruption of the outer mitochondrial membrane. One model suggests that the opening of the permeability transition pore (PTP) is the initial change that leads to cytochrome c release. PTP is thought to be a complex composed of many proteins including hexokinase, voltage dependent anion channel (VDAC), adenine nucleotide translocator (ANT) and cyclophilin D (CypD). Opening of this pore leads to equilibration of solutes up to 1.5 kDa across mitochondrial membranes. This results in depolarization and subsequent matrix swelling due to colloidosmotic pressure. The second model suggests an initial hyperpolarization due to inhibition of ATP/ADP exchange via VDAC as reason for matrix swelling. Other models propose formation of pores for cytochrome c release. Bax (and maybe also other proapoptotic members of the bcl-2 family) could form channels in the outer membrane or alternatively cause membrane instability that leads to cytochrome c release. It has also been proposed that Bax regulates the pore size of VDAC to allow cytochrome c translocation
A delicate mechanism of cytochrome c release involving only the permeabilization of the outer membrane is suggested by findings that Bcl-2 family proteins can form pores in liposomes or artificial membranes. Structural studies of Bcl-xL43 revealed that the protein had a similar arrangement to the translocation domain of diphtheria toxin and of the colicins. Upon exposure to acid pH, the transmembrane domain of diphtheria toxin is inserted into the lipid bilayer and forms an ion channel, followed by translocation of the catalytic domain through the lipid bilayer.44,45 Similarly, recombinant Bcl-xL lacking the C-terminal hydrophobic region (Bcl-xLΔC) was shown to form an ion channel in lipid lilayers at pH 4–5.46 Subsequently it was shown that Bcl-2ΔC also exhibited an ion channel activity and released Cl− from liposomes at pH below 5.5.47 Whether ion channel activity could account for the physiological role of Bcl-2 proteins remains an open question as (a) it is unlikely that mitochondria would be exposed to such extreme acidic pH and (b) Bcl-2 proteins regulate cytochrome c release from isolated mitochondria at neutral pH (and presumably in cells).
Subsequently the structure of Bid, a pro-apoptotic member of the Bcl-2 family, was determined. Its striking similarity to that of Bcl-xLΔC suggested that Bcl-2 family proteins all interact with lipid bilayers in a similar manner.48,49 As expected, Bid induced Cl− efflux from liposomes and exhibited ion channel activity in planar bilayers, with both activities facilitated by acidic pH.50 More recently, there have been reports showing that recombinant Bid releases encapsulated proteins (cytochrome c and trypsin)51 as well as carboxyfluorescein (CF)52 from liposomes at neutral pH, without involving other proteins. In addition, the pro-apoptotic Bcl-2 protein, Bax, released CF from liposomes at neutral pH although this activity was increased at lower pH.53 Significantly, the release of CF by BaxΔC was inhibited by addition of Bcl-2ΔC at neutral pH,53 raising the possibility that Bcl-2ΔC exerts its effect without forming an ion channel.
Kudla et al.52 found that Bid did not form ion channels, but ruptured the planar bilayer. In addition, although BaxΔC could form ion channels at low concentrations, at higher levels it also ruptured the lipid bilayer. In vitro translated full length Bax has also been found to rupture the membrane.54 In summary, Bcl-2 family proteins can alter the permeability of lipid bilayers composed of various lipids, though negatively charged lipids were preferred,46,47,50 suggesting that they can interact with the mitochondrial outer membrane in the same manner. Future studies utilizing the lipid composition reflecting that of the outer membrane of mitochondria at physiological pH will enhance our understanding of this process.
Although evidence has been published that Bax and Bid can permeabilize membranes on their own, Tsujimoto and colleagues55 found that VDAC (voltage dependent anion channel), the most abundant protein located in the outer membrane of mitochondria was necessary for Bax to exert its effect. Bax also co-immunoprecipitated with VDAC from isolated mitochondria and in cells undergoing apoptosis.55 Liposomes became permeable to sucrose upon incorporation of VDAC and this permeability was enhanced by recombinant full length Bax. The Bax effect was inhibitable by Bcl-xL.55 Further, electrophysiological studies showed that Bax protein, together with VDAC in the planar bilayer, formed a novel ion channel larger than VDAC or Bax alone.56 Permeability to cytochrome c was also demonstrated in the VDAC-loaded liposomes and in the planar bilayer systems in response to recombinant Bak or Bax.55,56
Thus, Bcl-2 family proteins, as their structures suggest, can interact with lipid bilayers, making them permeable to ions and small molecules and possibly to proteins as well. Alternatively, Bax, Bak and Bcl-xL may require proteins such as VDAC to release cytochrome c from mitochondria. Future studies including more comprehensive reconstitution will reveal whether Bcl-2 proteins regulate mitochondrial outer membrane proteins during apoptosis or act via direct pore formation.
Another suggested mechanism of apoptotic cytochrome c release is the mitochondrial permeability transition followed by large amplitude swelling that ultimately results in mechanical disruption of the outer membrane. Since a loss of mitochondrial membrane potential can be detected in apoptotic cells,57,58 permeability transition (PT) was proposed as a possible mechanism for cytochrome c release. Permeability transition is an event described in the early days of mitochondrial research since some time after isolation, the mitochondria lose their ability for oxidative phosphorylation and eventually swell. It was then discovered that to promote mitochondrial survival, calcium must be excluded from the buffers. Now we know that the permeability transition can be induced by calcium, phosphate, low membrane potential or atractyloside (ATR) and inhibited by high membrane potential, adenine nucleotides, magnesium, bongkrekic acid and cyclosporin A. Until recently no physiological consequence was attributed to this event in cells, although regulation of intracellular calcium concentration, solute exchange between mitochondria and cytosol or even a role in heat generation, by analogy to brown adipose tissue were proposed.59
To investigate whether permeability transition could lead to release of cytochrome c and so be involved in apoptosis regulation, Kroemer and colleagues60,61 treated isolated mitochondria with PT inducers and have shown that PT inducers (e.g. atractyloside, tBHP, CCCP) were sufficient to trigger apoptotic changes in added nuclei and that this was inhibitable by Bcl-2. Later however Marzo and colleagues62,63 showed that active caspases can induce PT pore opening and release of proteins from the mitochondrial intermembrane space suggesting the possibility that PT is a secondary event caused by active caspases that might work as an amplification loop. Very recently, a mass spectroscopic approach to identify proteins released from mitochondria upon ATR induced PT revealed that this treatment led to a general disruption of membranes and to the release of mitochondrial matrix proteins and even proteins from contaminating peroxisomes and lysosomes.64 These studies on artificial induction of PT proved that PT is sufficient to induce apoptosis, however since any mechanism of cytochrome c release will initiate an apoptotic response in cells, it has to be determined which events are necessary under physiological conditions to release cytochrome c. Since cytochrome c release in apoptotic cells is mediated by proapoptotic Bcl-2 proteins, the mechanism of cytochrome c release should be investigated using the effector proteins that mediate this event in vivo.
Taking this approach, Marzo et al .65 showed release of cytochrome c and loss of Δψm in isolated mitochondria treated with Bax and inhibition of Bax-dependent events by CsA and bongkrekic acid. However, it was not investigated whether Δψm loss preceded cytochrome c release or was a late consequence of this event. Bax and the adenine nucleotide transporter (ANT) were proposed to physically interact, and a yeast strain lacking all three ANT genes did not die from Bax transfection, so ANT was proposed to be necessary for Bax to kill yeast. It was not shown however, whether Bax could mediate cytochrome c release from yeast mitochondria independently of ANT, and we don't know yet how Bax mediated death of yeast relates to cytochrome c release and apoptosis in multicellular organisms.
Most results obtained using yeast as a model system seem to show that an intact mitochondrial energy metabolism is necessary for Bax toxicity. Inner membrane proteins like ANT, respiratory chain components and a working ATP synthase were reported to be needed 65,66,67,68 whereas the involvement of VDAC and even the occurrence of cytochrome c release gave controversial results. 55,67,69,70 Since cytochrome c release is not convincingly associated with Bax toxicity in yeast this organism might not be a suitable model for apoptotic cytochrome c release.
Pastorino et al. 71 showed that moderate concentrations of Bax can release intermembrane space proteins without loss of membrane potential whereas high Bax concentrations induced a permeability transition as indicated by loss of membane potential and large amplitude swelling.71 They proposed that transient permeability transition pore openings may lead to release of proteins from the mitochondrial intermembrane space but that a permanent opening is needed for a measurable depolarization and large amplitude swelling.71 However the authors failed to explain or even discuss how intermembrane space proteins could be released without large amplitude swelling, an event that seems to be required for the rupture of the outer membrane due to PT. A possible explanation may be that low concentrations of Bax are already sufficient for cytochrome c release to occur via a PT independent mechanism but that high Bax may lead to inner membrane damage by its pore forming or membrane destabilizing capabilities (see above).
Another hypothesis for cytochrome c release involves an initial hyperpolarization of mitochondria. Altered mitochondrial metabolism is proposed to result in an increased negative charge across the mitochondrial inner membrane. This increased stress would then lead to an influx of water to the matrix followed by rupturing of the outer membrane and subsequent cytochrome c release. However it is not clear whether this would result in an immediate loss of Δψm in cells, since mitoplasts can maintain membrane potential even though the outer membrane is fragmented.
Using the fluorescent dye rhodamine 123, an increase in Δψm (hyperpolarization) followed by a dramatic loss of Δψm (depolarization) was detected early during apoptosis of FL5.12 cells induced to die by growth factor withdrawal.13 The re-addition of growth factor rescued cells with hyperpolarized mitochondria but did not rescue cells that had lost Δψm. These results suggested that hyperpolarization occurred before commitment to apoptosis, and therefore probably before cytochrome c release. Both hyperpolarization and apoptosis were inhibited by Bcl-xL.13 In recent reports, Vander Heiden et al. 72,73 demonstrated that during growth factor withdrawal induced apoptosis the outer membrane became impermeable to complex anions preventing the transport of ATP/ADP while the transfer of ATP/ADP across the inner membrane via the ANT was unaffected. This change in permeability was similar to that observed in planar lipids containing VDAC and indicated that VDAC may take on a closed state during apoptosis. After the ADP stores in the matrix have been used up, it is possible that continuing action of the electron transport chain may lead to an increase in mitochondrial Δψm. The data presented also supported an anti-apoptotic role for Bcl-2 and Bcl-xL by maintaining ATP/ADP exchange across the outer membrane thereby preventing the increase in Δψm.
Matsuyama et al.68 recently demonstrated that oligomycin, an inhibitor of ATP synthase could inhibit apoptosis. ATP synthase utilizes the mitochondrial Δψm to form ATP from ADP and Phosphate. Although ATP synthase can operate in reverse, under normal physiological conditions oligomycin would be expected to reduce the demand on the mitochondrial Δψm resulting in hyperpolarization.68 If hyperpolarization were the mechanism for cytochrome c release oligomycin would be thought to facilitate the release of cytochrome c and induce apoptosis. Indeed others have shown that oligomycin can induce apoptosis, however this appears to take some time, rather than by direct hyperpolarization of the mitochondria.74 More recently, Matsuyama et al.75 demonstrated that although it induced a mild increase in Δψm, oligomycin appears to prevent the extent of hyperpolarization observed during staurosporine induced death. 75 This study also reported that an increase in matrix pH also preceded cytochrome c release. It is therefore possible that hyperpolarization is a consequence of altered metabolic events and that these events rather than hyperpolarization per se are responsible for cytochrome c release. Neither hyperpolarization nor an increase in pH of the mitochondrial matrix have been reported in isolated mitochondria treated with pro-apoptotic Bcl-2 family members and there is no hypothesis as to why an increased pH in the mitochondria would lead to cytochrome c release. Therefore we conclude that these changes may reflect metabolic alterations in dying cells but are not strictly required for cytochrome c release.