Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells (original) (raw)
Introduction
Neuroendocrine differentiation (NE) has been demonstrated in a variety of carcinomas arising in various tissues. Prostatic carcinoma, non-small-cell lung cancer, breast cancer, gastric carcinoma, and colorectal carcinoma are some of the tumors in which neuroendocrine differentiation has been described and suggested as an indicator of poor prognosis. However, the molecular and cellular mechanisms controlling NE differentiation are only partially understood.
Prostate cancer, one of the leading threats to men's health, is dependent on the androgens in the early stages. Consequently, androgen ablation therapies may, at this time, cause a tumor to regress. Nevertheless, these treatments do not prevent evolution to an androgen-independent stage, for which there is currently no successful therapy.1 Therefore, an understanding of what drives the progression to androgen independence is critical. It is well established that androgen-independence is associated with tumor enrichment in cell phenotypes, for which apoptosis inhibition rather than enhanced proliferation is the main feature.2,3 Malignant neuroendocrine (NE) cells represent one of such phenotypes.4
NE cells are fully differentiated cells that share structural, functional and metabolic properties with neurons.5 They are a normal component of both the developing and the mature prostate epithelium,4,5,6 although their origin and functional role is as yet, poorly elucidated. According to the existing hypotheses, NE cells are either derived from undifferentiated basal cells of the prostatic epithelium,7 or have a neurogenic origin.8 It is suggested that by releasing a variety of neurosecretory products, such as parathyroid hormone related peptides, neurotensin, serotonin, calcitonin and bombesin-related peptides,4,5,6 these cells participate in the regulation of normal development and secretory activity of the prostate in the endocrine/paracrine fashion.
NE cells lack nuclear androgen receptors9,10 and thus represent an androgen insensitive cell phenotype in the prostate. Expanding their population beyond normal proportions as a result of malignant transformation of epithelial/basal cells developing dormant NE features, is a common characteristic of prostate cancer progression.4 Tumor enrichment in NE cells and the consequent increase in neurosecretory products with growth-promoting properties, contribute to increases in malignancy and reduce responsiveness to androgen ablation therapy.4,5,6,11
An additional factor that greatly enhances the malignant potential associated with NE differentiation, is that of non-proliferating NE tumor cells escaping apoptotic cell death.3 The mechanisms of NE cells' apoptotis-resistance are still obscure, although existing evidence suggests that they are not associated with the common anti-apoptotic oncoprotein Bcl-2,12 but are rather triggered by recently discovered survival proteins, such as survivin13 and clusterin.14 It is obvious that further studies of all aspects of NE cells' apoptotis-resistance are imperative for the development of new therapeutic options in the treatment of advanced prostate cancer.
In a recent study15 we demonstrated that the apoptosis-resistance of androgen-dependent LNCaP (Lymph Node Carcinoma of the Prostate16) prostate cancer epithelial cells, associated with Bcl-2 overexpression, results in a complex rearrangement of the whole intracellular Ca2+ homeostasis. The characteristics of this rearrangement are reduced filling of the endoplasmic reticulum (ER) Ca2+ store, a decreased expression of key ER Ca2+ handling proteins and a substantial down-regulation of store-operated Ca2+ current (ISOC). Bcl-2 overexpression is known to transform androgen-dependent LNCaP cells into an androgen-independent phenotype.2 Since LNCaP cells can be differentiated into NE cells17,18,19 – which are qualitatively different from the Bcl-2-mediated mechanisms of apoptosis-resistance and androgen independence and as it is also known that Ca2+ signaling plays a key role in proliferation and apoptosis,20,21 we therefore sought in the present study, to examine the type and manner of NE differentiation-induced alterations in Ca2+ homeostasis. Our results show for the first time that NE differentiation of prostate cancer epithelial cells causes significant modifications in Ca2+ homeostasis, which in their major hallmark features (i.e. the reduced filling of the ER Ca2+ store, the decreased expression of both endolemmal SERCA 2b Ca2+ ATPase and the luminal Ca2+ binding/storage chaperone calreticulin, as well as a substantial ISOC down-regulation) mimicked those associated with Bcl-2 overexpression. This would suggest the existence of common sites for Ca2+-dependent triggering of various anti-apoptotic pathways in androgen-independent prostate cancer. We also show that NE-differentiated LNCaP cells become resistant to thapsigargin- (TG) and TNF-_α_-induced apoptosis.
Results
LNCaP cells rapidly acquire NE characteristics including the cessation of mitotic activity, the development of neuritic processes and an increased expression of neuron-specific enolase (NSE), under either pharmacological stimulation increasing intracellular cAMP levels, or under physiological stimulations using interleukins or long-term androgen deprivation.17,18,19,22 In a previous work23 we presented evidence of LNCaP cells' NE differentiation in response to interventions that increase intracellular cAMP ([cAMP]in) levels. In the present study, we have used both [cAMP]in elevation and long-term androgen deprivation by means of charcoal stripped culture medium, in order to induce NE differentiation of LNCaP cells. The functional results obtained on NE-differentiated LNCaP cells were compared with regular androgen-dependent LNCaP cells, which served as a control.
Neuroendocrine differentiation decreases both ER free Ca2+ concentration and store-operated calcium entry
In terms of basic morphological NE characteristics, the two NE-differentiated cell types were similar to those described previously.23 Culturing LNCaP cells for 4 days with a cAMP permeant analog (dibutyryl cAMP, Bt2cAMP 1 mM) plus a phosphodiesterase inhibitor (isobutylmethylxanthine, IBMX 100 _μ_M) or with an androgen-deprived culture medium, induced NE differentiation. In order to comprehend the possible alterations in overall Ca2+ homeostasis induced by NE differentiation of LNCaP cells, we used a widely used experimental protocol, which allows the dissection of intracellular Ca2+ release from Ca2+ entry in a single cell fluorometric measurement of cytosolic free Ca2+ concentration ([Ca2+]c). This protocol consisted in Fura-2-loaded cell exposure to a SERCA pump inhibitor, TG, in nominally Ca2+-free extracellular saline, which should thus reveal the liberation of only intracellularly stored Ca2+, with the subsequent re-addition of extracellular Ca2+ to initiate the influx via activated store-operated Ca2+ entry (SOCE). Figure 1a compares the results of such an experiment in a representative control in LNCaP cell (ctrl-LNCaP) and LNCaP cells differentiated into NE phenotype by either [cAMP]in elevation (NE-cAMP-LNCaP, pre-treatment for 96 h with 1 mM Bt2cAMP plus 100 _μ_M IBMX), or by androgen deprivation (NE-Andr(-)-LNCaP). An inspection of [Ca2+]c traces in Figure 1a suggests that NE differentiation, irrespectively of how it was achieved, did not affect basal [Ca2+]c level ([Ca2+]c, basal), but decreases both the amounts of liberated Ca2+ ([Ca2+]c, lib) and of Ca2+ influx ([Ca2+]c, SOCE). The quantification of basal [Ca2+]c and its rise due to store-operated Ca2+ influx, in up to 240 cells of each type (Figure 1b), confirmed this conclusion and provided numerical values for [Ca2+]c, basal of 52±1.7, 49±1.5 and 45±1.6 nM and for [Ca2+]c, SOCE of 757±22, 462±34 and 509±21 nM in the ctrl-LNCaP, NE-cAMP-LNCaP and NE-Andr(-)-LNCaP cells, respectively. The reductions in [Ca2+]c, SOCE were insignificant after 24 h of initial NE differentiating treatment (irrespective of the nature of the treatment), and became maximal after 96 h when NE morphological features were fully acquired.23
Figure 1
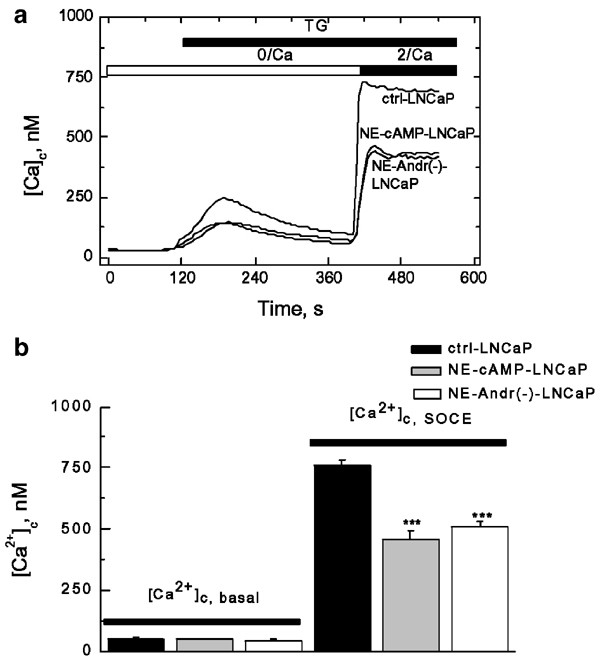
NE differentiation of LNCaP cells reduces thapsigargin-induced Ca2+ influx, but does not affect basal Ca2+. (a) Representative experiments on TG- (1 _μ_M) induced changes in cytoplasmic Ca2+ concentration ([Ca2+]c, measured using fura-2 fluorescence) in LNCaP control cells (ctrl-LNCaP) and NE-differentiated LNCaP cells by [cAMP]in elevation (NE-cAMP-LNCaP) or androgen deprivation (NE-Andr(−)-LNCaP); periods of TG application and extracellular Ca2+ elevation from 0 (0/Ca) to 2 mM (2/Ca) are marked by horizontal bars; see text for details. (b) Quantification of basal cytoplasmic Ca2+ concentration ([Ca2+]c,basal) and store-operated Ca2+ entry ([Ca2+]c,SOCE) in the control LNCaP cells (ctrl-LNCaP) and two types of NE-differentiated LNCaP cells (NE-cAMP-LNCaP and NE-Andr(−)-LNCaP); mean±s.e.m., _n_=173–239
As [Ca2+]c, lib apparently becomes notably lower in NE-differentiated cells too (Figure 1a), this suggests that their ER Ca2+ store content ([Ca2+]ER) is lower than that of the control. However, the magnitude of the apparent [Ca2+]c, lib signal may not directly reflect [Ca2+]ER, as it can be substantially affected by ER-independent Ca2+ uptake mechanisms, of the mitochondrial type in particular, which may undergo upregulation during NE differentiation. In order to obtain more direct evidence on the filling status of ER Ca2+ store, we performed two additional series of experiments. In the first, we used cell exposure to ionomycin (IM, 1 _μ_M) – a Ca2+ ionophore that causes complete ER Ca2+ store depletion when incorporated into membranes.24 This was carried out in nominally Ca2+-free extracellular saline, supplemented with the mitochondrial inhibitors, oligomycin (40 _μ_M) and rotenon (20 _μ_M),15 in order to provide conditions under which IM-induced [Ca2+]c, lib would more fully reflect the actual [Ca2+]ER. In the second series, we directly monitored [Ca2+]ER in digitonin-permeabilized cells by means of the compartmentalized fluorescent dye, Mag-fura 2-AM.15
The summaries of the experiments on the effects of NE differentiation on ER Ca2+ store content are presented in Figure 2. As is evident from individual traces (Figure 2a), the IM-induced [Ca2+]c, lib under inhibited mitochondrial uptake, is about 50% lower in NE-differentiated LNCaP cells than in control cells (i.e., 200±9 nM, _n_=176 versus 413±29 nM, _n_=197). This suggests that their ER Ca2+ store content would also be lower by approximately the same percentage. This conclusion was generally confirmed by direct Mag-fura 2-AM [Ca2+]ER measurements, although they provided a somewhat lower [Ca2+]ER decrease (−35%) following 96 h of NE differentiation (from 540±35 _μ_M, _n_=25 to 350±40 _μ_M, _n_=29, Figure 2c), whereas 24 h appeared to be insufficient to produce a statistically significant [Ca2+]ER decrease (Figure 2b).
Figure 2
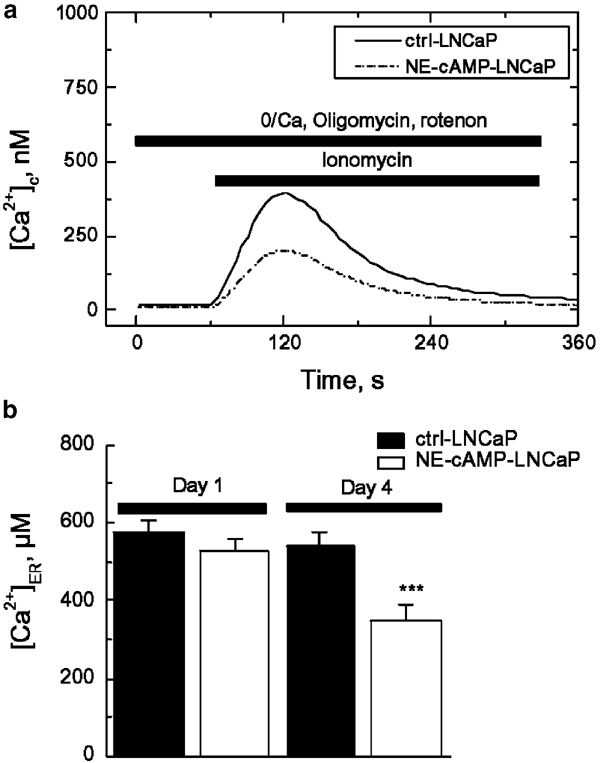
NE differentiation of LNCaP cells reduces ER Ca2+ content. (a) Representative experiments on ionomycin (1 _μ_M) induced Ca2+ liberation ([Ca2+]c, which was measured using fura-2 fluorescence) from the ER in the LNCaP control cells (ctrl-LNCaP) and NE-differentiated LNCaP cells (NE-cAMP-LNCaP) bathed in nominally Ca2+-free extracellular saline (0/Ca) supplemented with the mitochondrial inhibitors oligomycin (40 _μ_M) and rotenon (20 _μ_M); mean±s.e.m., _n_=197–176; see text for details. (b) The quantification of the ER intraluminal Ca2+ concentration ([Ca2+]ER, measured in digitonin-permeabilized cells based on Mag-fura 2AM fluorescence) in the LNCaP control cells (ctrl-LNCaP) and LNCaP cells subjected to NE-differentiating treatment (NE-cAMP-LNCaP) with Bt2cAMP (1 mM) plus IBMX (100 _μ_M) 1 and 4 days after treatment initiation; mean±s.e.m., _n_=25–29; see text for details
NE differentiation reduces a store-operated current
To determine whether or not the reduction in SOCE following NE differentiation, detected in fluorometric experiments, is indeed associated with a decrease in the store-operated membrane Ca2+ current (ISOC), we proceeded to record the whole cell-patch clamp of this current both in the control and in the NE-differentiated LNCaP cells. In this series of experiments, we dialyzed cells with a high concentration of BAPTA, a highly potent Ca2+ chelator, which rapidly binds Ca2+ passively leaking from the ER, thereby causing its depletion, thereby activating ISOC.25
Figure 3a shows that progression of the dialysis with 10 mM BAPTA containing intracellular solution, caused the development of inward ISOC in all three LNCaP cell types: control and NE-differentiated both by means of [cAMP]in elevation or androgen deprivation. The development of these currents followed comparable time courses, but the maximal densities, reached in about 4 min following the establishment of a whole-cell configuration, were different (Figure 3a). The I-Vs of fully developed currents in all cell types showed a strong inward rectification and reversal potential at around +40 mV, which is typical of ISOC (Figure 3b). The quantification of maximal current density at 100 mV and 10 mM Ca2+ as a charge carrier, showed that it is the largest of the control LNCaP cells (1.4±0.2 pA/pF, _n_=12), which however decreases to 0.8±0.1 pA/pF and 0.6±0.1 pA/pF (_n_=8–9) following 96 h of NE differentiation by [cAMP]in elevation or androgen deprivation, respectively (Figure 3c). 24 h of differentiation were insufficient to reveal statistically significant differences in current densities.
Figure 3
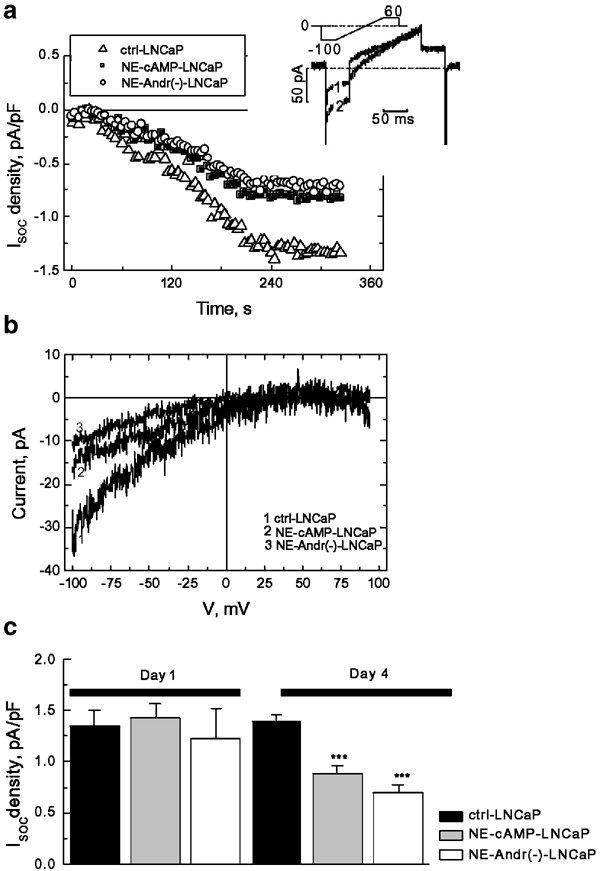
NE differentiation of LNCaP cells reduces store-operated Ca2+ current. (a) Representative time courses of store-operated Ca2+ current development (_I_SOC, normalized to membrane capacitance to yield current density) in response to 10 mM BAPTA infusion in the control LNCaP cells (ctrl-LNCaP) and NE-differentiated LNCaP cells by [cAMP]in elevation (NE-cAMP-LNCaP) or androgen deprivation (NE-Andr(−)-LNCaP) at 10 mM [Ca2+]out and _V_m=−100 mV. The inset shows an example of the original baseline (1) and fully developed (2) currents together with voltage-clamp protocol. (b) Representative I_–_V relationships of _I_SOC in control (ctrl-LNCaP) and NE-differentiated (NE-cAMP-LNCaP and NE-Andr(−)-LNCaP) cells derived from currents in response to voltage ramps. (c) The quantification of maximal _I_SOC density at −100 mV in the control LNCaP cells (ctrl-LNCaP) and LNCaP cells subjected to NE-differentiating treatments by [cAMP]in elevation (NE-cAMP-LNCaP) or androgen deprivation (NE-Andr(−)-LNCaP) 1 and 4 days after treatment initiation; mean±s.e.m., _n_=12–8–9, respectively; see text for details
Thus, the data on direct ISOC recordings indicate that the reduction in SOCE observed in NE-differentiated LNCaP cells by fluorometric means, is associated with a decrease in the density of store-operated current.
NE differentiation decreases SERCA 2b and calreticulin expression
One of the mechanisms by which NE differentiation may decrease the Ca2+ filling status of the ER, is by affecting the expression of ER-specific Ca2+-handling proteins, such as the endolemmal SERCA pump and/or the luminal Ca2+ binding/storage chaperone, calreticulin. We therefore compared the expression of these proteins in both control and NE-differentiated cells, using a semi-quantitative Western blot analysis. We analyzed the expression pattern of three other proteins, NSE, Bcl-2 and Bcl-xl. NSE, a neuroendocrine marker, is important in the appreciation of NE differentiation, whereas the assessment of common anti-apoptotic oncoproteins Bcl-2 and Bcl-xl, may provide a clue about the mechanisms of NE cells' apoptotis-resistance.
Figure 4a shows that androgen deprivation as well as the [cAMP]in elevation, increase NSE expression (1.84 versus 1.74-fold respectively). This was also accompanied by a greater decrease in levels of SERCA 2b (0.52 versus 0.69-fold) and calreticulin (0.48 versus 0.61-fold). Such expression patterns pointed strongly to the fact that a reduction in both proteins, SERCA pump and calreticulin, constitute a major factor determining decreased ER Ca2+ filling status during NE differentiation. Surprisingly, androgen deprivation and [cAMP]in elevation resulted in a notable decrease in Bcl-2 levels (to 0.65 and 0.49 of the control value respectively) and in the Bcl-xl level (to 0.8 and 0.6, Figure 4b), suggesting that an anti-apoptotic oncoprotein-independent mechanism contributes to NE cells' apoptotis-resistance.
Figure 4
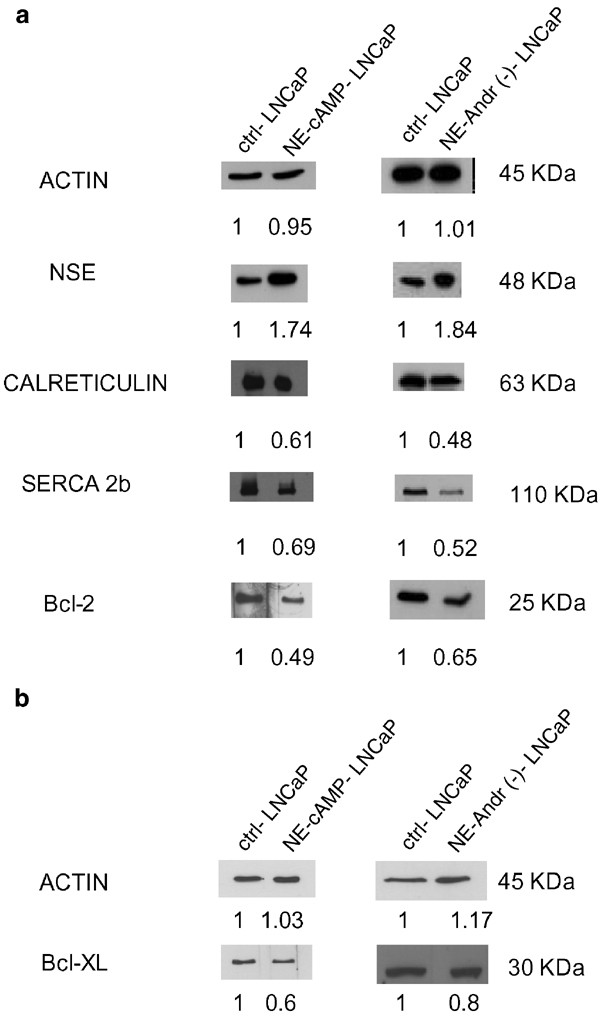
NE differentiation of LNCaP cells alters the expression of the ER Ca2+-handling proteins. Semiquantitative Western blots (after 30 _μ_g per well of total protein extraction for Bt2cAMP+IBMX treatment and 50 _μ_g per well of total protein extraction for androgen deprivation) showing (a) increased expression of the NE marker NSE and underexpression of the endolemmal SERCA 2b Ca2+ pump, the luminal Ca2+ binding/storage chaperone calreticulin, and the antiapoptotic Bcl-2 and (b) Bcl-xl proteins in LNCaP cells subjected to 96 h of NE-differentiating treatments by [cAMP]in elevation (NE-cAMP-LNCaP) or androgen deprivation (NE-Andr(−)-LNCaP) compared to the controls (ctrl-LNCaP). Actin expression was assessed to ensure identical experimental conditions. All stains in (a) were performed on a single membrane at the same time, except in the case of the Bcl-xl protein, where we have added the corresponding actin control (b)
Androgen deprivation confers resistance to thapsigargin and TNF-_α_-induced apoptosis
Possible alterations in the NE-differentiated LNCaP cells' potential to resist pro-apoptotic stimuli were assessed using TG, which has previously been shown to be an effective experimental inducer of apoptosis in LNCaP cells, through an ER depletion mechanism,26 as well as by using a physiological pro-apoptotic factor, namely TNF-α, acting via a surface membrane death receptor.27 These experiments were conducted on NE-differentiated LNCaP cells by androgen deprivation, as this regimen is more physiological and more relevant from a clinical perspective.
Figure 5a shows that, in control LNCaP cells, treatment with TG for 48 h induced apoptosis in a dose dependent manner, with a maximal percentage of apoptotic cells observed for 1 _μ_M TG (26±2.1%). Following NE differentiation, the fraction of apoptotic cells was reduced approximately 2-fold both in the absence of TG, as well as for each TG dose (Figure 5a), thereby suggesting enhanced apoptotis resistance. The treatment of NE differentiated cells with TG for 72 h did not increase the fraction of apoptotic cells (data not shown), consequently suggesting that the apoptosis in differentiated cells was not delayed.
Figure 5
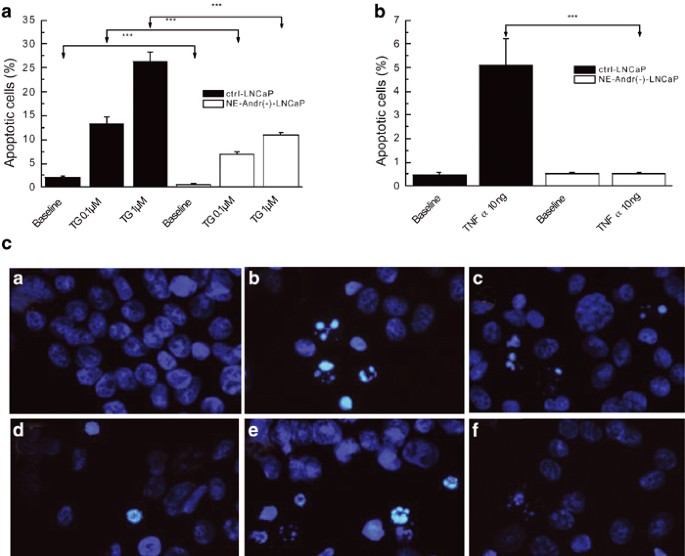
NE differentiation of LNCaP cells enhances their antiapoptotic potential. (a) Bar graph showing the much lower percentage of apoptotic NE-differentiated LNCaP cells (after 96 h of androgen deprivation, NE-Andr(−)-LNCaP) in response to two concentrations of TG (0.1 and 1 _μ_M) for 48 h, compared to the control LNCaP cells (ctrl-LNCaP); ‘***’ denotes P<0.01. (b) Same as in (a), but for 10 ng/ml TNF-α. (c) Illustration of the typical apoptotic features with the Hoechst technique in each condition. (a–c): ctrl-LNCaP cells (a), LNCaP cells with TG 1 _μ_M (b) or TNF-α 10 ng/ml (c); (d–f): NE-Andr(−)-LNCaP cells (d), NE-Andr(−)-LNCaP cells with TG 1 _μ_M (e) or TNF-α 10 ng/ml (f)
Qualitatively similar results were obtained with TNF-α (10 ng). This pro-apoptotic factor increased control LNCaP cells' apoptosis nearly 10-fold (from 0.5±0.1 to 5.1±1.4%), but appeared to be completely ineffective in inducing apoptosis of NE-differentiated LNCaP cells (Figure 5b). The typical apoptotic features induced by treatment with TG or TNF-α are shown in Figure 5c. Apoptosis resistance was also detected by the TUNEL technique (Figure 6a), thus demonstrating the decrease in LNCaP differentiated cells' apoptosis. Moreover, our results are confirmed by the DNA ladder technique (Figure 6b).
Figure 6
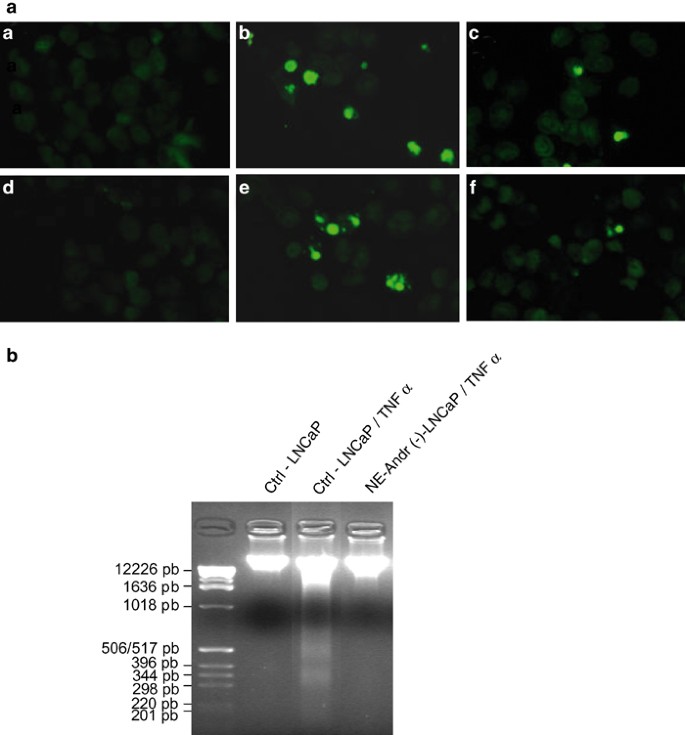
Apoptosis detection in control and differentiated LNCaP prostate cells using the TUNEL and the DNA ladder techniques. (a) The TUNEL technique, a and d showing undifferentiated control cells and differentiated LNCaP cells, respectively; (b, c) represent undifferentiated cells treated for 48 h with TG 1 _μ_M or TNF-α 10 ng/ml, respectively; (e, f) represent the same as in (b, c), but with differentiated cells. The DNA ladder technique (b) shows a reduction in the DNA fragmentation of differentiated LNCaP cells under TNF-α 10 ng/ml treatment for 48 h
Discussion
In the present study, we report two major findings characterizing NE differentiation in prostate cancer. Firstly, we describe the effect on Ca2+ homeostasis manifested through reduced filling of ER Ca2+ store, due to the under-expression of major Ca2+-handling proteins, SERCA 2b and calreticulin and the downregulation of store-operated Ca2+ entry. Secondly, we demonstrate an enhanced resistance to apoptosis, which is not related to Bcl-2 and Bcl-xl, but, we believe, is a consequence of altered Ca2+ homeostasis.
Ca2+ homeostasis and apoptosis
The involvement of Ca2+-dependent mechanisms in the induction and regulation of apoptosis is now generally accepted. Although interrelated, depending on the site of action, these mechanisms can be roughly subdivided into mitochondrial, cytoplasmic and ER-dependent types. Mitochondria may respond to high-matrix Ca2+. This might be accumulated as a result of enhanced sequestration, either through a transition in permeability allowing the release of the following: Ca2+, some matrix components as well as an apoptogenic factor, cytochrome c, or perhaps through an enhanced production of reactive oxygen species.28 A rise in cytosolic free Ca2+, which has long been considered as a primary death signal, leads to the activation of the calpain family of Ca2+-dependent proteases participating in apoptosis,29 and to the increase in the activity of some caspases.30 Ca2+ homeostasis normally plays a pivotal role in the folding and processing of newly synthesized proteins as well as in maintaining the overall physiological state of the cell. When this decreases due to the depletion of ER Ca2+ stores, a stress response is activated, leading to growth arrest and cell death.31 It is therefore possible, that disrupting intracellular calcium homeostasis, by the alteration of the way in which proteins regulating calcium homeostasis are expressed and function, will affect the cell's sensitivity to apoptosis. For example, it was shown that the over-expression of the intraluminal ER protein, calreticulin, increased cell sensitivity to TG- and staurosporine-induced apoptosis and, conversely, that cells lacking calreticulin showed considerable resistance to drug-induced apoptosis.32
Androgen ablation therapy in prostatic adenocarcinoma induces an involution of prostate tissue mainly through the enhancement of cellular apoptosis,33 which necessarily involves a pro-apoptotic decrease in the ER Ca2+ filling status. A fraction of malignant cells withstanding such therapy would emerge as new populations of apoptotis-resistant cells, for which underfilled ER represents another natural level of functional equilibrium. The enrichment of the prostate with such cell phenotypes eventually causes virtually all tumors to relapse into an androgen-independent, more aggressively growing type.
Apoptotis resistance in prostate cancer following androgen ablation therapy may evolve through two major pathways: the overexpression of the antiapoptotic Bcl-2 protein2 or Bcl-2-independent NE differentiation.3 In a recent study, we characterized Ca2+ homeostasis in a model system of LNCaP prostate cancer epithelial cells transfected with Bcl-2,15 and showed that the Bcl-2-conferred apoptotis resistance on these cells is associated with reduced ER Ca2+ content and the substantial downregulation of _I_SOC. We also showed that reduced ER filling was a consequence of enhanced leakage and the lower expression of the key ER Ca2+-handling proteins SERCA 2b and calreticulin, whereas diminished _I_SOC most probably reflected an adaptive decrease in the number of functional channels to the long-term reduction in the ER Ca2+ content.15
The results of our present work show that alterations in Ca2+ homeostasis induced by the NE differentiation of LNCaP cells, which are schematically represented in Figure 7, basically mimic those of Bcl-2-overexpressing LNCaP cells, in terms of such hallmark features as an underfilled ER Ca2+ store, lower levels of ER SERCA 2b and calreticulin, and reduced _I_SOC.
Figure 7
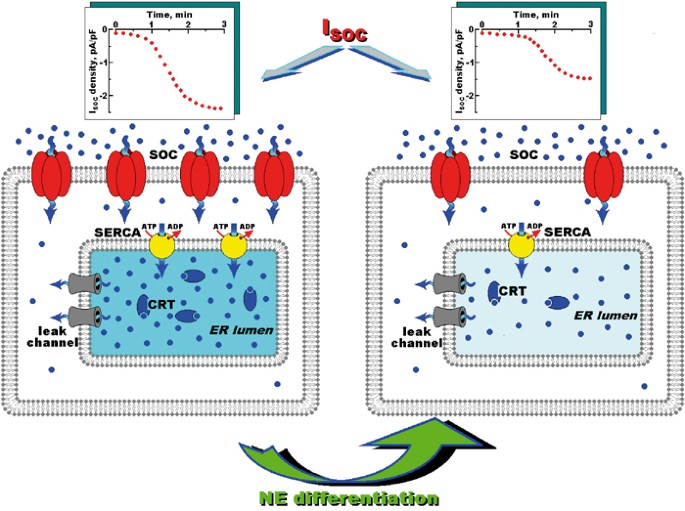
Schematic diagram showing the major effects of prostate cancer cells neuroendocrine differentiation on Ca2+ homeostasis. The left-hand panel presents the control conditions characterized by basal expression of the ER leak channels, the SERCA pump, the intraluminal CRT, and the plasma membrane SOCs, which when taken together, result in the background ER Ca2+ concentration and store-operated Ca2+ entry (_I_SOC, see upper left graph) typical of control, androgen-dependent prostate cancer epithelial cells. Neuroendocrine differentiation (right-hand panel) results in an ER intraluminal Ca2+ concentration which had been lowered through the downregulated expression of SERCA pump and CRT, and decreased store-operated Ca2+ entry, which was most probably associated with the diminished density of functional SOCs
Mechanisms of NE cells apoptotis resistance
Apoptosis is an extremely rare event in prostate cancer cells with neuroendocrine features in primary, metastatic, and recurrent disease.3 Recent studies suggest that the apoptotis resistance of NE cells may be related to overexpression of new survival proteins, survivin13 and/or clusterin.34,14 The first of these is a member of the inhibitor of apoptosis (IAP) family with direct caspase-3 and caspase-7 inhibitory action,35 whereas the second represents multifunctional glycoprotein, commonly involved in the transport of lipoproteins, the inhibition of complement-mediated cell lysis, and the modulation of cell–cell interactions.36 The antiapoptotic mechanisms of the latter remain to be clarified. Survivin is highly expressed in various common human cancers,37,38,39 but not in normal tissues, and clusterin expression is strongly enhanced in tissues undergoing apoptosis.40
The fact that LNCaP cells undergo similar alterations in Ca2+ homeostasis, irrespective of whether their apoptotis resistance was enhanced by Bcl-2 overexpression or as a result of NE differentiation, suggests that these alterations create a generally favorable environment for the functioning of mechanisms counteracting apoptosis. These alterations are therefore a necessary prerequisite for the cell's ability to successfully withstand proapoptotic stimuli. Our previous work showed that NE differentiation of LNCaP cells is also characterized by a strong overexpression of voltage-gated T-type Ca2+ channels,23 which seem to be involved in the formation of neuronal-like morphological features (i.e., neurite outgrowth). Whether or not these channels contribute to the enhanced antiapoptotic potential of NE cells is not yet clear.
Implications for androgen-independent prostate cancer
Apoptosis is essential in maintaining tissue homeostasis. The acquisition of a resistance to apoptosis plays a pivotal role in tumor genesis by disrupting the balance between cell proliferation and cell destruction, and also by allowing cancer cells to escape radiation and chemotherapy. Androgen-independent prostate cancer is characterized by tumor enrichment in apoptosis-resistant cell phenotypes, which, although differing in specific antiapoptotic mechanisms, nevertheless shares the same basic changes in intracellular Ca2+ homeostasis. It would therefore seem that targeting the key players involved in its maintenance, in an attempt to enhance the proapoptotic potential of malignant cells, may prove to be a useful strategy in the treatment of advanced prostate cancer.
In the present work, we identify three molecular entities, the expression of which undergoes significant changes (reduction) during the NE differentiation of prostate cancer epithelial cells – calreticulin, SERCA 2b, and store-operated channel (SOC). The molecular origin of SOCs is still unknown, but there is strong evidence that the most likely candidate proteins belong to the transient receptor potential (TRP) channel family.41,42,43 Thus, the identification of the specific TRPs involved in endogenous _I_SOC in prostate cancer cells acquires potentially great practical importance, as they may represent appropriate targets for influencing the apoptotic status of NE cells in advanced, androgen-independent prostate cancer.
Materials and Methods
Cell culture
The human prostatic carcinoma cell line LNCaP was obtained from the American Type Culture Collection (Rockville, MD, USA). Cells were grown in RPMI 1640 with 10% decomplemented fetal bovine serum (FBS, Deutcher, Brumath, France) at 37°C, in a humidified atmosphere containing 5% CO2. The medium was supplemented with 300 _μ_g/ml L-glutamine, 100 U/ml penicillin, and 100 _μ_g/ml streptomycin. Tissue culture media and supplements were obtained from Life Technologies (Cergy Pontoise, France). Cells were seeded in 75 cm2 flasks and the growth medium was renewed every other day.
Prior to fluorescence measurements, the cells were removed from the culture flasks with 0.05% trypsin (Life Technologies, Cergy Pontoise, France), and cultured on glass cover slips in the same culture medium. Cells were then used 2 days after trypsinization.
Charcoal-stripped medium
The tube containing charcoal 10% (w/v) and FBS was agitated for 16 h at 4°C. Following 1 h's centrifugation at 10 000 g and 4°C, the supernatant was collected and centrifuged again for 30 min at 27 000 g. The resultant supernatant was filtrated through 0.22 _μ_m filters. Before use, the charcoal-stripped FBS was decomplemented for 30 min at 56°C.
Fluorescence [Ca2+]c measurements and fluorescence [Ca2+]ER measurements
For both the cytoplasmic and the ER Ca2+ imaging, the detailed procedure has been described previously.15,23
Electrophysiology and solutions
Membrane currents in LNCaP cells were recorded in the whole-cell configuration of the patch-clamp technique, using a computer-controlled EPC-9 amplifier (HEKA Electronic, Germany), as described previously.44 Patch pipettes were made on a PIP-5 puller (HEKA Electronic, Germany) from borosilicate glass capillaries (WPI, USA). The resistance of the pipettes filled with the basic intracellular pipette solution (in mM): CsCl – 120, MgCl2 – 3, BAPTA – 10, HEPES – 10, pH – 7.3 (adjusted with Cs(OH)) varied between 4 and 6 MΩ.
The composition of the regular extracellular bath solution was (in mM): NaCl – 120, KCl – 5, CaCl2 – 2, MgCl2 – 2, glucose – 5, HEPES – 10, pH – 7.3 (adjusted with Na(OH)). The high-Ca2+, Na+-free extracellular solution used for store-operated Ca2+ current recordings contained (in mM): TEA-Cl – 120, CaCl2 – 10, glucose – 5, HEPES – 10, pH – 7.3 (adjusted with TEA(OH)). External solutions were changed using a multibarrel puffing micropipette with common outflow, positioned in close proximity to the cell under investigation. During the experiment, the cell was continuously superfused with the solution via a puffing pipette to reduce possible artifacts related to the switch from static to moving solution and vice versa. Complete external solution exchange was achieved in less than 1 s.
Western blot analysis
Cells were lysed in an ice-cold buffer (pH 7.4) containing (in mM) 20 HEPES, 50 NaCl, 10 EDTA, 1 EGTA, 1 PMSF, 1% NP40, and a mixture of protease inhibitors. After 1 h on ice, the lysates were homogenized and centrifuged at 1500 rpm for 10 min at 4°C. The resulting supernatants were stored at −80°C until use. Samples were fractionated in a Laemmli-type SDS-PAGE (16%). The proteins were then transferred onto a nitrocellulose membrane using a semi-dry electroblotter (Bio-Rad). After transfer, the membrane was cut into thin strips that were further processed for immunodetection. The strips were blocked in TNT (15 mM Tris buffer (pH 8), 140 mM NaCl, 0.05% Tween 20, 3% skimmed dry milk for 30 min at room temperature), washed in TNT three times, then incubated with mouse monoclonal antibodies for 1 h at room temperature. After three washes in TNT, the strips were treated with the corresponding horseradish peroxidase-linked secondary antibodies (Zymed Laboratoires Inc., San Francisco, CA, USA), for 1 h. After washes in TNT without milk, the strips were processed for chemiluminescent detection using Supersignal West Pico chemiluminescent substrate (Pierce chemical Co., Rockford, IL, USA), according to the manufacturer's instructions. Blots were then exposed to X-Omat AR films (Eastman Kodak Co., Rochester, NY, USA). Blot intensity was quantified by scanning densitometry (Bio-Rad).
Determination of apoptosis
The Hoechst technique
The level of apoptosis was estimated from the number of apoptotic bodies visualized by Hoechst staining. The percentage of apoptotic cells was determined by counting at least 500 cells in random fields (the detailed procedure has been described previously.26
The TUNEL technique
Apoptosis was also detected by the terminal deoxynucleotide transferase-mediated dUTP-biotin nick-end labellingterminal deoxynucleotide transferase-mediated dUTP-biotin nick-end labelling technique (TUNEL) using an apoptosis detection kit (Borhringer Mannheim).
DNA fragmentation analysis
Cells (2 × 106 cells per sample) were lysed in an extraction buffer (50 mM Tris pH 7.5, 10 mM EDTA, 1% sodium dodecyl sulfate (SDS), and 200 _μ_g/ml proteinase K, 50 _μ_g/ml RNase A) and incubated at 37°C for 6 h. DNA was subsequently extracted with phenol/chloroform/isoamyl alcohol (25 : 24 : 1) and then precipitated with isopropyl alcohol, pelleted, and resuspended in TE with 20 _μ_g/ml Rnase. The DNA was quantitated spectroscopically, and 10 _μ_g of DNA was analyzed for fragmentation on a 1.5% agarose gel. The gel was stained with ethidium bromide and visualized using a UV light source.
Reagents and chemicals
All chemicals were from Sigma (l'Isle d'Abeau, France), except for fura-2/AM and thapsigargin, which were purchased from France Biochem (Meudon, France).
Data analysis and statistics
Each experiment was repeated several times. The data were analyzed using PulseFit (HEKA Electrinics, Germany) and Origin 5.0 (Microcal Software Inc., Northampton, MA, USA). Results were expressed as mean±standard error of the mean (S.E.M.). Statistical analysis was performed using the Student's _t_-test and ANOVA tests, followed by Tukey–Kramer post-tests (P<0.05 considered significant).
Abbreviations
NE:
neuroendocrine
LNCaP:
lymph node carcinoma of the prostate
ER:
endoplasmic reticulum
I SOC :
store-operated Ca2+ current
TG:
thapsigargin
NSE:
neuron-specific enolase
Bt2cAMP:
dibutyryl cAMP
IBMX:
isobutylmethylxanthine
SOCE:
store-operated Ca2+ entry
ctrl-LNCaP:
control LNCaP cell
NE-cAMP-LNCaP:
LNCaP cells differentiated into NE phenotype by [cAMP]in elevation
NE-Andr(−)-LNCaP:
LNCaP cells differentiated into NE phenotype by androgen deprivation
TUNEL technique:
terminal deoxynucleotide transferase-mediated dUTP-biotin nick-end labelling
IAP:
inhibitor of apoptosis
CRT:
calreticulin
References
- Feldman BJ and Feldman D (2001) The development of androgen-independent prostate cancer. Nat. Rev. Cancer 1: 34–45
Article CAS Google Scholar - Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS and Buttyan R (1995) Overexpression of Bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 55: 4438–4445
CAS PubMed Google Scholar - Fixemer T, Remberger K and Bonkhoff H (2002) Apoptosis resistance of neuroendocrine phenotypes in prostatic adenocarcinoma. Prostate 53: 118–123
Article Google Scholar - Abrahamsson PA (1999) Neuroendocrine cells in tumour growth of the prostate. Endocr. Relat. Cancer 6: 503–519
Article CAS Google Scholar - di Sant'Agnese PA (1998) Neuroendocrine cells of the prostate and neuroendocrine differentiation in prostatic carcinoma: a review of morphologic aspects. Urology 51: 121–124
Article CAS Google Scholar - di Sant'Agnese PA (1992) Neuroendocrine differentiation in carcinoma of the prostate. Diagnostic, prognostic, and therapeutic implications. Cancer 70: 254–268
Article CAS Google Scholar - Bonkhoff H and Remberger K (1996) Differentiation pathways and histogenetic aspects of normal and abnormal prostatic growth: a stem cell model. Prostate 28: 98–106
Article CAS Google Scholar - Aumuller G, Leonhardt M, Janssen M, Konrad L, Bjartell A and Abrahamssonn PA (1999) Neurogenic origin of human prostate endocrine cells. Urology 53: 1041–1048
Article CAS Google Scholar - Krijnen JL, Janssen PJ, Ruizeveld de Winter JA, van Krimpen H, Schroder FH and van der Kwast TH (1993) Do neuroendocrine cells in human prostate cancer express androgen receptor? Histochemistry 100: 393–398
Article CAS Google Scholar - Bonkhoff and H (2001) Neuroendocrine differentiation in human prostate cancer. Morphogenesis, proliferation and androgen receptor status. Ann. Oncol. 12: S141–S144
Article Google Scholar - Ito T, Yamamoto S, Ohno Y, Namiki K, Aizawa T, Akiyama A and Tachibana M (2001) Up-regulation of neuroendocrine differentiation in prostate cancer after androgen deprivation therapy, degree and androgen independence. Oncol. Rep. 8: 1221–1224
CAS PubMed Google Scholar - Xue Y, Verhofstad A, Lange W, Smedts F, Debruyne F, de la Rosette J and Schalken J (1997) Prostatic neuroendocrine cells have a unique keratin expression pattern and do not express Bcl-2: cell kinetic features of neuroendocrine cells in the human prostate. Am. J. Pathol. 151: 1759–1765
CAS PubMed PubMed Central Google Scholar - Xing N, Qian J, Bostwick D, Bergstralh E and Young CY (2001) Neuroendocrine cells in human prostate over-express the anti-apoptosis protein survivin. Prostate 48: 7–15
Article CAS Google Scholar - July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL and Gleave ME (2002) Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate 50: 179–188
Article CAS Google Scholar - Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F and Prevarskaya N (2002) Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 1: 169–179
Article CAS Google Scholar - Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA and Murphy GP (1983) LNCaP model of human prostatic carcinoma. Cancer Res. 43: 1809–1818
CAS PubMed Google Scholar - Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, Ha MJ, Tsokos M, Sheahan MD and Nguyen P (1994) Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc. Natl. Acad. Sci. USA 91: 5330–5334
Article CAS Google Scholar - Burchardt T, Burchardt M, Chen MW, Cao Y, de la Taille A, Shabsigh A, Hayek O, Dorai T and Buttyan R (1999) Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J. Urol. 162: 1800–1805
Article CAS Google Scholar - Zelivianski S, Verni M, Moore C, Kondrikov D, Taylor R and Lin MF (2001) Multipathways for transdifferentiation of human prostate cancer cells into neuroendocrine-like phenotype. Biochim. Biophys. Acta 1539: 28–43
Article CAS Google Scholar - Berridge MJ, Bootman MD and Lipp P (1998) Calcium – a life and death signal. Nature 395: 645–648
Article CAS Google Scholar - McConkey DJ and Orrenius S (1997) The role of calcium in the regulation of apoptosis. Biochem. Biophys. Res. Commun. 239: 357–366
Article CAS Google Scholar - Deeble PD, Murphy DJ, Parsons SJ and Cox ME (2001) Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol. Cell. Biol. 21: 8471–8482
Article CAS Google Scholar - Mariot P, Vanoverberghe K, Lalevée N, Rossier MF and Prevarskaya N (2002) Overexpression of an alpha IH (Cav 3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J. Biol. Chem. 277: 10824–10833
Article CAS Google Scholar - Huang Jr Y and Putney JW (1998) Relationship between intracellular calcium store depletion and calcium release-activated calcium current in a mast cell line (RBL-1). J. Biol. Chem. 273: 19554–19559
Article CAS Google Scholar - Hoth M and Penner R (1993) Release-activated calcium current in rat mast cells. J. Physiol. 465: 359–386
Article CAS Google Scholar - Skryma R, Mariot P, Bourhis XL, Coppenolle FV, Shuba Y, Vanden Abeele F, Legrand G, Humez S, Boilly B and Prevarskaya N (2000) Store depletion and store-operated Ca2+ current in human prostate cancer LNCaP cells: involvement in apoptosis. J. Physiol. 527: 71–83
Article CAS Google Scholar - Ashkenazi A and Dixit VM (1998) Death receptors: signaling and modulation. Science 281: 1305–1308
Article CAS Google Scholar - Kass GE and Orrenius S (1999) Calcium signaling and cytotoxicity. Environ. Health Perspect. 107: 25–35
CAS PubMed PubMed Central Google Scholar - Mathiasen IS, Sergeev IN, Sergeev L, Bastholm F, Elling AW, Norman and Jaattela M (2002) Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J. Biol. Chem. 277: 30738–30745
Article CAS Google Scholar - Juin P, Pelletier M, Oliver L, Tremblais K, Gregoire M, Meflah K and Vallette FM (1998) Induction of a caspase-3-like activity by calcium in normal cytosolic extracts triggers nuclear apoptosis in a cell-free system. J. Biol. Chem. 273: 17559–17564
Article CAS Google Scholar - Paschen W and Doutheil J (1999) Disturbance of endoplasmic reticulum functions: a key mechanism underlying cell damage? Acta Neurochir. Suppl. (Wien) 73: 1–5
CAS Google Scholar - Nakamura K, Bossy-Wetzel E, Burns K, Fadel MP, Lozyk M, Goping IS, Opas M, Bleackley RC, Green DR and Michalak M (2000) Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150: 731–740
Article CAS Google Scholar - Montironi R, Pomante R, Diamanti L and Magi-Galluzzi C (1998) Apoptosis in prostatic adenocarcinoma following complete androgen ablation. Urol. Int. 60: 25–39
Article Google Scholar - Lee C, Janulis L, Ilio K, Shah A, Park I, Kim S, Cryns V, Pins M and Bergan R (2000) In vitro models of prostate apoptosis: clusterin as an antiapoptotic mediator. Prostate Suppl. 9: 21–24
Article CAS Google Scholar - Yamamoto T and Tanigawa N (2001) The role of survivin as a new target of diagnosis and treatment in human cancer. Med. Electron. Microsc. 34: 207–212
Article CAS Google Scholar - Koch-Brandt C and Morgans C (1996) Clusterin: a role in cell survival in the face of apoptosis? Prog. Mol. Subcell. Biol. 16: 130–149
Article CAS Google Scholar - Takai N, Miyazaki T, Nishida M, Nasu K and Miyakawa I (2002) Expression of survivin is associated with malignant potential in epithelial ovarian carcinoma. Int. J. Mol. Med. 10: 211–216
CAS PubMed Google Scholar - Nasu S, Yagihashi A, Izawa A, Saito K, Asanuma K, Nakamura N, Kobayashi D, Okazaki M and Watanabe N (2002) Survivin mRNA expression in patients with breast cancer. Anticancer Res. 22: 1839–1843
CAS PubMed Google Scholar - Kajiwara Y, Yamasaki F, Hama S, Yahara K, Yoshioka H, Sugiyama K, Arita K and Kurisu K (2003) Expression of survivin in astrocytic tumors. Cancer 97: 1077–1083
Article Google Scholar - Viard I, Wehrli P, Jornot L, Bullani R, Vechietti JL, Schifferli JA, Tschopp J and French LE (1999) Clusterin gene expression mediates resistance to apoptotic cell death induced by heat shock and oxidative stress. J. Invest. Dermatol. 112: 290–296
Article CAS Google Scholar - Clapham DE, Runnels LW and Strubing C (2001) The TRP ion channel family. Nat. Rev. Neurosci. 2: 387–396
Article CAS Google Scholar - Minke B and Cook B (2002) TRP channel proteins and signal transduction. Physiol. Rev. 82: 429–472
Article CAS Google Scholar - Venkatachalam K, van Rossum DB, Patterson RL, Ma HT and Gill DL (2002) The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol. 4: E263–E272
Article CAS Google Scholar - Vanden Abeele F, Roudbaraki M, Shuba Y, Skryma R and Prevarskaya N (2003) Store-operated Ca2+ current in prostate cancer epithelial cells. Role of endogenous Ca2+ transporter type 1. J. Biol. Chem. 278: 15381–15389
Article CAS Google Scholar
Acknowledgements
This work was supported by grants from INSERM, La Ligue Nationale Contre le Cancer and l'ARC (France), Association pour la Recherche sur les Tumeurs de la Prostate (ARTP), Fondation pour la Recherche Médicale (FRM), and INTAS-99-01248. YM Shuba was supported by INSERM and the French Ministry of Science.
Author information
Author notes
- K Vanoverberghe and F Vanden Abeele: These authors contributed equally to this work and should be considered co-first authors.
Authors and Affiliations
- Laboratoire de Physiologie Cellulaire, INSERM EMI 0228, Université des Sciences et Technologies de Lille, Bât. SN3, Villeneuve d'Ascq, 59655, France
K Vanoverberghe, F Vanden Abeele, P Mariot, G Lepage, M Roudbaraki, J L Bonnal, B Mauroy, Y Shuba, R Skryma & N Prevarskaya - Bogomoletz Institute of Physiology NASU, Bogomoletz Str., 4, Kiev, 01024, Ukraine
Y Shuba
Authors
- K Vanoverberghe
You can also search for this author inPubMed Google Scholar - F Vanden Abeele
You can also search for this author inPubMed Google Scholar - P Mariot
You can also search for this author inPubMed Google Scholar - G Lepage
You can also search for this author inPubMed Google Scholar - M Roudbaraki
You can also search for this author inPubMed Google Scholar - J L Bonnal
You can also search for this author inPubMed Google Scholar - B Mauroy
You can also search for this author inPubMed Google Scholar - Y Shuba
You can also search for this author inPubMed Google Scholar - R Skryma
You can also search for this author inPubMed Google Scholar - N Prevarskaya
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toN Prevarskaya.
Additional information
Edited by JA Cidlowski
Rights and permissions
About this article
Cite this article
Vanoverberghe, K., Vanden Abeele, F., Mariot, P. et al. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells.Cell Death Differ 11, 321–330 (2004). https://doi.org/10.1038/sj.cdd.4401375
- Received: 09 May 2003
- Revised: 13 October 2003
- Accepted: 06 November 2003
- Published: 19 December 2003
- Issue Date: 01 March 2004
- DOI: https://doi.org/10.1038/sj.cdd.4401375