Lack of involvement of mitochondrial factors in caspase activation in a Drosophila cell-free system (original) (raw)
Introduction
Apoptosis, a type of programmed cell death, is a highly regulated process used to remove unwanted cells in metazoans. Apoptosis is required for normal development, maintenance of tissue homeostasis, and regulation of certain disease states.1, 2 The key components of the machinery that carries out this evolutionarily conserved process are caspases. Caspases are a group of cysteine proteases that are synthesized as zymogens, but upon receiving an apoptotic signal, the inactive caspases usually undergo proteolytic processing to generate the active form.3
In mammals, there are two well-defined pathways that lead to caspase activation, the extrinsic, and intrinsic pathways.4 In the intrinsic pathway, a death signal causes the release of cytochrome c from the intermembrane space of mitochondria into the cytosol, where it binds to the adaptor molecule Apaf-1, causing Apaf-1 to undergo a conformational change that allows it to bind ATP and procaspase-9.5 Procaspase-9 is then activated by dimerization, and activated caspase-9 cleaves effector caspases such as caspase-3 and caspase-7, which cleave key substrates in the cell, leading to death.6 In addition to cytochrome c, several other proapoptotic factors are known to be released from the intermembrane space of mammalian cells following apoptotic stimulation, including Smac/DIABLO, AIF, endonuclease G, and HtrA2/Omi. These factors serve to inhibit inhibitor of apoptosis (IAP) proteins, which normally function to inhibit active caspases, and also can induce caspase-independent death.4
The role of cytochrome c in the mammalian intrinsic apoptotic pathway is well documented. Addition of cytochrome c and ATP to cytosolic extract is sufficient to cause activation of caspase-3,7 and injection of cytochrome c into mammalian cells triggers caspase activation and apoptosis.8 Furthermore, cells derived from mouse embryos lacking cytochrome c are severely impaired in the intrinsic death pathway.9
In Drosophila, a genetic screen led to the identification of three genes that are involved in developmentally programmed apoptosis: reaper (rpr), head involution defective (hid), and grim.10 The products of each of these genes induce apoptosis via a pathway that requires caspase action. These proapoptotic proteins, along with two other proteins, Sickle11 and Jafrac2,12 bind to the Drosophila IAP protein DIAP1, and one mechanism by which these proteins promote apoptosis is by disrupting DIAP1–caspase interactions.13 Seven Drosophila caspases have been identified, and four of these, DCP-1, DrICE, Dredd, and DRONC, have been implicated in the regulation of apoptosis.14, 15, 16, 17 In addition, a Drosophila homolog of Apaf-1/CED4, called DARK/Dapaf-1/HAC-1,18 has been identified and shown to be required for DRONC activation.19
Although the apoptotic machinery in _Drosophil_a appears to be superficially similar to that of mammals, the role of mitochondrial factors in Drosophila apoptosis is unclear. One study found that in Drosophila SL2 cells, induction of apoptosis by overexpression of Reaper or treatment with staurosporine or cycloheximide led to the release of cytochrome c into the cytosol.20 However, Varkey et al.21 reported that caspase activity or overexpression of Reaper or Grim did not lead to cytochrome c release, but instead led to altered cytochrome c display. Zimmermann et al.22 demonstrated that after treatment with UV, cycloheximide, or actinomycin D, cytochrome c remained in the mitochondria and when cytochrome c expression was reduced by RNAi, Drosophila cells showed no increased resistance to apoptosis induced by Reaper or Grim. Overexpression of cytochrome c in Drosophila BG2 cells or addition of recombinant cytochrome c to cytosolic BG2 extract did not lead to increased caspase activation or apoptosis.23 Genetic analysis of cytochrome c mutants has also led to the conclusion that it is not necessary for caspase activation in Drosophila. Prepupae that were heterozygous for P-element insertions in either or both of the two cytochrome c alleles in Drosophila, dc3, and dc4, or heterozygous for a deficiency that removed both alleles, had reduced levels of cytochrome c but still had normal levels of caspase activity.
Although these previous studies demonstrated that cytochrome c is not necessary for caspase activation, they failed to conclusively rule out whether it can play a role in this process. For example, Dorstyn et al.24 also found that incubation of cytosolic extract from BG2 cells with cytochrome c and ATP caused DRONC to become associated with a large molecular weight complex reminiscent of the apoptosome in mammals, suggesting that cytochrome c might be capable of causing caspase activation.24 In addition, the potential role of other mitochondrial factors besides cytochrome c in caspase activation has not been investigated in Drosophila. One study has shown that Reaper localizes to mitochondria via its GH3 domain, and the authors hypothesized that Reaper may induce the release of mitochondrial factors that influence caspase activation,25 but evidence in this area is lacking.
In this study, we have investigated the importance of cytochrome c and other mitochondrial factors in caspase activation in Drosophila S2 cell extracts. We report that cytochrome c and other mitochondrial factors are not required for, nor do they appear to influence, caspase activation in S2 cytosolic extracts. Our results instead indicate that one or more of the cytosolic proteins Hid, Reaper, and Grim are both necessary and sufficient to induce caspase activation in S2 cells due to their abilities to inhibit the interaction between DIAP1 and DRONC and to trigger DIAP1 degradation.
Results
The majority of previous studies that have examined cytochrome c release from mitochondria in Drosophila cells have concluded that cytochrome c is not released from Drosophila mitochondria following apoptotic stimuli.21, 22, 23, 24 To examine this, we treated S2 cells with UV and at various times after UV treatment samples were harvested and assayed for cytochrome c and DrICE in the mitochondrial (P10) and cytosolic (S100) fractions by immunoblot analysis. Similar to previous reports, cytochrome c was only detected in the P10 fraction even though caspase activity and processed DrICE were observed in the S100 fraction (Figure 1a). In contrast, when human 293 cells were UV-irradiated, cytochrome c was released into the S100 fraction, as is known to occur in mammalian cells (Figure 1b). Cytochrome c release into the S100 fraction was also not observed in S2 cells when apoptosis was induced by overexpression of Hid, Reaper, or Grim (Figure S1).
Figure 1
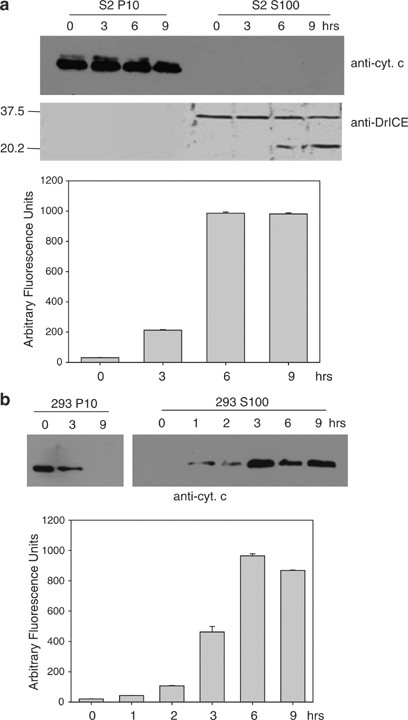
Cytochrome c is not released from S2 cell mitochondria during UV-induced apoptosis. (a) Drosophila S2 cells were UV irradiated and at the indicated times the P10 and S100 fractions were isolated and analyzed by immunoblotting with anti-cytochrome c and anti-DrICE antibodies (above). Caspase activity was measured in S100 from UV-treated cells using DEVD-afc as substrate (below). (b) Human 293 cells were UV irradiated and at the indicated times the P10 and S100 fractions were isolated and analyzed by immunoblotting with anti-cytochrome c antibody (above). Caspase activity was measured in S100 from UV-treated cells using DEVD-afc as substrate (below)
Previous studies have also suggested that cytochrome c is not required for Drosophila apoptosis, because cells having low levels of cytochrome c still undergo normal apoptosis.22, 23 We verified these results using RNA-mediated interference (RNAi) to reduce endogenous cytochrome c levels. Drosophila encodes two cytochrome c proteins, DC3, and DC4, and it has been shown that RNAi using dc4 sequences inhibits expression of both genes.22 Immunoblot analysis showed a significant reduction in cytochrome c protein levels in the P10 fraction of S2 cells treated with dc4 dsRNA compared to untreated cells (Figure 2a). Depletion of cytochrome c by RNAi had no effect on apoptosis stimulated by UV or by overexpression of Hid, Rpr, or Grim (Figure S2), and DrICE processing was normal in UV-treated S2 cells in which cytochrome c was depleted (Figure 2b). In addition, when S100 from 1 × 108 S2 cells that were treated with cytochrome c dsRNA and UV irradiated was mixed with S100 from 2 × 109 untreated cells, the levels of DrICE processing and DEVD-ase activity in the untreated lysate were normal (Figure 2c–e). To verify that the active DrICE detected in the immunoblot in Figure 2c was not from the UV-treated S100 lysate, we analyzed the same amount of UV-treated S100 that was added to untreated lysate in Figure 2c by immunoblotting and found that DrICE was undetectable in these samples under these conditions (data not shown).
Figure 2
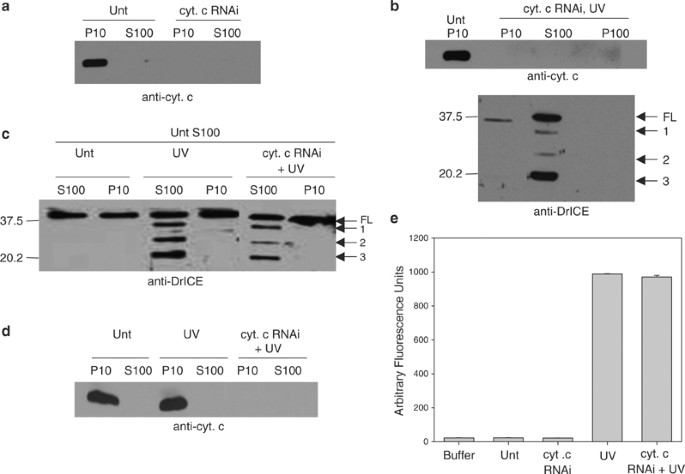
Cytochrome c is not necessary for caspase activation in S2 cells. (a) S2 cells were left untreated (Unt) or were treated with dsRNA corresponding to cytochrome c and 72 h later the indicated fractions were isolated and analyzed by immunoblotting with anti-cytochrome c antibody. (b) S2 cells were treated with cytochrome c dsRNA and 72 h later the cells were UV irradiated and analyzed 4 h later by immunoblotting with anti-DrICE antibody. Full-length (FL) DrICE was processed to species 1 (lacking the 28 amino-acid prodomain), 2 (prodomain plus large subunit), and 3 (the p19 large subunit). (c) S100 from 2 × 109 untreated S2 cells was incubated with P10 or S100 isolated from 1 × 108 S2 cells that were untreated, UV irradiated, or UV irradiated and treated with cytochrome c dsRNA. Following the incubation, an immunoblot was performed using anti-DrICE antibody. (d) Anti-cytochrome c immunoblot of the lysates in panel c that were incubated with untreated S100. (e) Measurement of caspase activity (using Ac-DEVD-afc as substrate) in S100 isolated from S2 cells that were treated with cytochrome c dsRNA and UV irradiated
The ability of S100 from UV-irradiated cells to cause caspase activation in S100 from untreated cells could have been due to either cytosolic proteins (including activated caspases) or to mitochondrial factors (other than cytochrome c) that had been released into the cytosol following UV treatment, or both. To eliminate active caspases from the UV-treated lysate, we performed RNAi to the apical caspase DRONC, followed by UV irradiation. Following DRONC and cytochrome c RNAi, DRONC and cytochrome c protein levels were undetectable by immunoblotting (data not shown) and DrICE was not processed in UV-irradiated cells (Figure 3a). Also, the dronc dsRNA-treated cells did not undergo apoptosis when UV irradiated (data not shown) and neither apical (IETD-ase) nor effector (DEVD-ase) caspase activity were detectable (Figure 3b). However, when S100 from DRONC RNAi, UV-irradiated cells was mixed with S100 from untreated cells, DrICE activation was still observed (Figure 4a, lane 3). This indicated that either some other cytosolic factor besides activated caspases or a mitochondrial factor that had been released into the cytosol was responsible for caspase activation. DrICE processing was not observed when untreated S100 was mixed with S100 obtained from cells that were not irradiated, regardless of whether cytochrome c, DRONC, or both had been depleted (Figure 4a, lanes 2, 4 and 5).
Figure 3
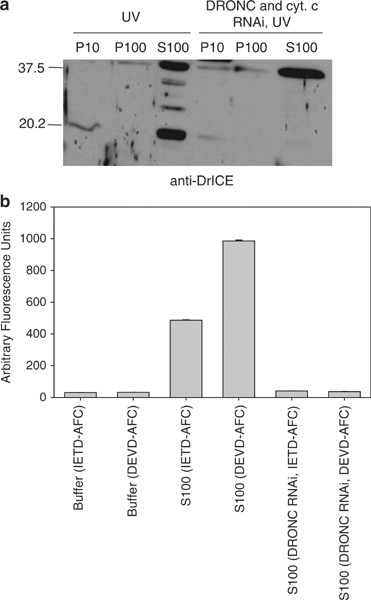
Silencing of the apical caspase DRONC eliminates DrICE processing and caspase activity. (a) S2 cells were treated with dsRNA corresponding to cytochrome c and DRONC and the indicated fractions were isolated and immunoblotted using anti-DriCE antibody. (b) Caspase activity was examined in S2 S100 that was treated with dronc dsRNA using Ac-IETD-afc or Ac-DEVD-afc as substrates
Figure 4
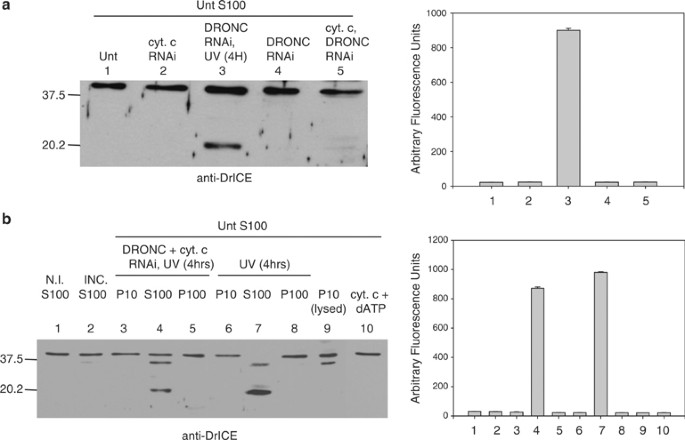
Mitochondrial factors are not able to induce caspase activation in S2 cell lysate. (a) S100 from 2 × 109 untreated S2 cells was incubated with S100 from 1 × 108 S2 cells that were untreated, treated with dronc and/or cytochrome c dsRNA, or treated with dronc dsRNA and UV irradiated. An immunoblot was performed for DrICE (left) and caspase activity was determined using Ac-DEVD-afc as substrate (right). (b) DrICE immunoblot (left) and caspase activity assay using DEVD-afc (right) of the following samples: lane 1, nonincubated S100 from untreated S2 cells; lane 2, S100 from untreated S2 cells that was incubated alone; lanes 3–8, S100 from untreated S2 cells that was incubated with the indicated fractions from S2 cells that were UV irradiated or treated with dronc and cytochrome c dsRNA and UV irradiated; lane 9, S100 from untreated cells that was incubated with lysed P10 fraction; lane 10, S100 from untreated cells that was incubated with cytochrome c protein and dATP
To test whether a mitochondrial factor was responsible, the mitochondrial (P10) fraction was isolated from untreated cells, and mitochondria were lysed by four freeze–thaw cycles. The resulting lysate was then incubated with S100 fraction from untreated cells. Significant levels of DrICE processing or DEVD-ase activity were not observed in S100 extract treated with lysed mitochondria (Figure 4b, lanes 9). However, when extract from lysed mitochondria obtained from S2 cells was added to S100 obtained from untreated human 293 cells, caspase activation was observed (Figure 5b, sample 5). If the S2 cell mitochondria were not lysed, they did not cause caspase activation in 293 cell S100 (Figure 5b, sample 3). Neither intact nor lysed mitochondria from 293 cells were able to activate caspases in S100 from untreated S2 cells (Figure 5a, samples 2 and 4). Also, addition of cytochrome c and dATP to S100 from untreated S2 cells did not lead to DrICE processing (Figure 4b, lanes 10 and Figure 5a, sample 8), but did cause caspase activation in 293 cell S100 (Figure 5b, sample 8). The cytochrome c that was released from S2 mitochondria by freeze–thawing was required for inducing caspase activation in S100 from 293 cells, since RNAi of cytochrome c eliminated the caspase activating ability of lysed P10 from S2 cells (Figure 5b, sample 7). These results indicate that not only cytochrome c but also other mitochondrial factors are unable to cause caspase activation in S2 cell S100.
Figure 5
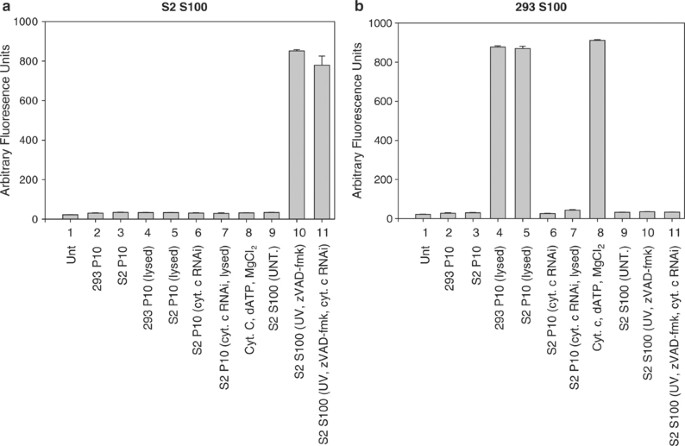
Lysed S2 cell mitochondria are capable of inducing caspase activation in S100 obtained from human cells but not in S100 from Drosophila cells. S100 lysate obtained from untreated 2 × 109 Drosophila S2 (a) or 5 × 106 human 293 (b) cells was incubated with either S2 or 293 P10, purified cytochrome c, or S100 from 1 × 108 UV-treated S2 cells treated with the caspase inhibitor zVAD-fmk and caspase activity was determined using DEVD-afc as substrate
The difference in requirement for mitochondrial factors in caspase activation between Drosophila and mammalian cells was further illustrated by the observation that when 293 S100 was incubated with S100 isolated from S2 cells that were UV irradiated and treated with the caspase inhibitor zVAD-fmk and cytochrome c dsRNA, caspase activation was not observed, but it was in the S2 S100 (Figure 5a, b, samples 11). The amount of zVAD-fmk used was sufficient to inhibit caspase activity in the S2 S100 (Figure 6, sample 9). To determine if the reason why the 293 S100 did not show caspase activity when incubated with the treated S2 S100 was due to the lack of mitochondria in the S100, 293 whole-cell lysate (containing both mitochondria and cytosolic factors) was used and the experiment was repeated. Caspases were activated in the 293 whole-cell lysate when it was incubated with the treated S2 S100 (Figure 6, sample 1), indicating that factors in the S2 S100 could only activate caspases in 293 cells if mitochondria were present.
Figure 6
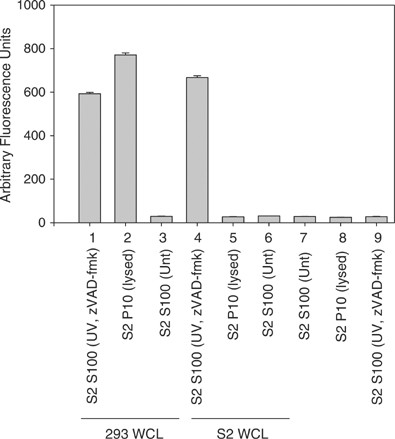
S100 from UV-treated S2 cells treated with caspase inhibitor is able to activate caspases in 293 or S2 whole-cell extract. S100 isolated from 1 × 108 S2 cells that were UV irradiated and treated with z-VAD-fmk was incubated alone (samples 7–9) or with 293 or S2 whole-cell lysate (WCL) and caspase activity was determined using DEVD-afc as substrate
These results indicated that one or more cytosolic factors, which did not include caspases, were responsible for caspase activation in S2 S100. Since Hid, Reaper and Grim are known to interrupt interactions between DIAP1 and caspases, we examined the effect on S2 cell viability when Hid, Rpr, and/or Grim were depleted by RNAi. The RNAi treatment greatly reduced the amount of mRNA for Hid, Rpr, and Grim as measured by RT-PCR (Figure S3). Cells that were treated with Hid, Reaper, or Grim dsRNA were protected from UV-induced apoptosis when compared to S2 cells that were treated with control CAT dsRNA (Figure 7a). S2 cells that were treated with Hid or Grim dsRNA were more protected than cells treated with Rpr dsRNA, while silencing both Hid and Grim enhanced protection. However, the best protection was observed when all three genes were silenced.
Figure 7
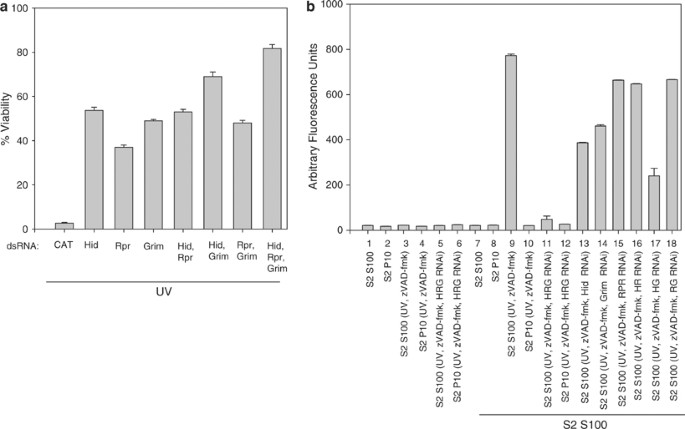
Silencing of Hid, Rpr, and/or Grim protects S2 cells against UV-induced apoptosis and inhibits caspase activation in S100. (a) S2 cells were treated with dsRNA corresponding to CAT (negative control), Hid, Rpr, Grim or combinations of Hid, Rpr and Grim. At 48 h after dsRNA addition, the cells were UV irradiated and 24 h later cell viability was determined. (b) S100 or P10 from 1 × 108 S2 cells that were UV irradiated and treated with dsRNA corresponding to Hid, Rpr, and Grim was incubated alone (samples 1–6) or with S100 from 2 × 109 untreated S2 cells (lanes 7–18). Caspase activity was determined using DEVD-afc as substrate
When S100 from untreated S2 cells was incubated with S2 S100 that was isolated from cells that were treated with UV, zVAD-fmk, and Hid, Reaper, and Grim dsRNA, caspase activation was significantly reduced compared to control cells not receiving dsRNA (Figure 7b, compare samples 9 and 11). Silencing Hid (sample 13) or Grim (sample 14) individually had more of an effect on caspase activity than silencing Rpr (sample 15) and silencing Hid and Grim together (sample 17) had more effect than Hid and Rpr (sample 16) or Grim and Rpr (sample 18) (Figure 7b). These results were consistent with the observed protection in Figure 7a.
When 293 whole-cell extract was incubated with S2 S100 isolated from cells that were UV irradiated and treated with zVAD-fmk, caspase activation was observed (Figures 6 and 8a, sample 3). However, when Hid, Rpr, and Grim were silenced, caspase activity was reduced (Figure 8a, sample 10). When the genes were silenced individually, more of an effect was observed when Reaper or Grim were silenced, while silencing Hid individually had little effect (Figure 8a, samples 4–6). However, silencing all three genes was more effective than silencing only Rpr and Grim (compare samples 9 and 10).
Figure 8
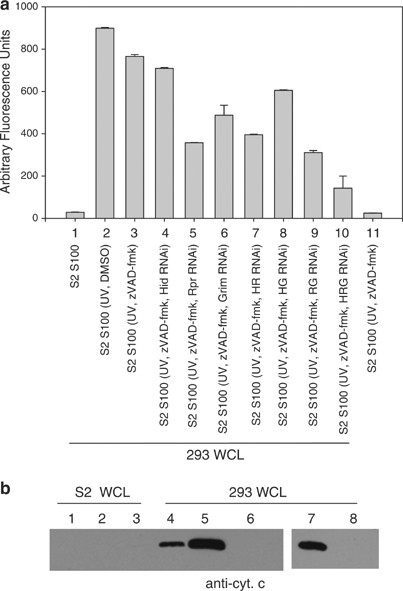
Silencing Hid, Rpr and Grim reduces the ability of S2 S100 to cause caspase activation in 293 whole-cell extract. (a) 293 whole-cell lysate (WCL) was incubated with S100 isolated from S2 cells that were UV irradiated and treated with zVAD-fmk and dsRNA corresponding to Hid, Reaper, or Grim. Caspase activity was determined using DEVD-afc as substrate. (b) Cytochrome c immunoblot of cytosolic fractions isolated from S2 or 293 WCL that had been incubated with S100 from S2 cells that were UV irradiated and treated with zVAD-fmk (lanes 1 and 4), UV irradiated (lanes 2 and 5), UV irradiated and treated with zVAD-fmk and Hid, Rpr and Grim dsRNA (lanes 3 and 6), UV irradiated and treated with Hid, Rpr and Grim dsRNA (lane 7), or untreated (lane 8)
To examine if the S2 S100 caused release of cytochrome c from the 293 cell mitochondria, 293 whole-cell lysates were incubated with treated S2 S100 and then separated into cytosolic and mitochondrial fractions and the cytosolic fractions were immunoblotted for cytochrome c (Figure 8b). 293 cell lysates treated with S100 obtained from S2 cells that were UV-irradiated (lane 5), UV-irradiated and treated with zVAD-fmk (lane 4), or UV-irradiated and treated with dsRNA to Hid, Rpr, and Grim (lane 7) all exhibited cytochrome c release in the cytosol, but when cells were UV irradiated and treated with both zVAD-fmk and dsRNA to Hid, Reaper and Grim, cytochrome c translocation to the cytosol did not occur (lane 6). Thus, either (1) active caspases or (2) Hid, Rpr, and Grim were sufficient to cause cytochrome c release from 293 cell mitochondria, but if caspases were inhibited and Hid, Rpr, and Grim were silenced, cytochrome c release was not observed.
The data in Figure 7 indicate that Hid, Rpr, and Grim (in particular Hid and Grim) are necessary for activation of caspases in S2 S100 extract. To determine if a DIAP1 antagonist was also sufficient to induce caspase activation in S2 cells, we used biotinylated peptides representing either the native or processed amino terminus of the Hid protein. As has been previously shown,26 a peptide representing the processed amino terminus of Hid lacking the initiating methionine residue (Hid peptide) bound to DIAP1, but a peptide including the initiating methionine residue (Met-Hid peptide) did not (Figure S4-a). Increasing concentrations of this Hid peptide induced DrICE cleavage and caspase activity in S100 extract from untreated S2 cells, while the Met-Hid peptide did not (Figure 9a, b). The Hid peptide was able to disrupt the interaction between DIAP1 and DRONC, as determined by immunoprecipitation of recombinant proteins, while the Met-Hid peptide did not (Figure S4-b). However the Hid peptide was unable to disrupt the interaction between DIAP1 and catalytically inactive DRONC (C318S) in which the active site cysteine was mutated to serine (Figure S4-b), supporting previous observations that suggest that the binding contacts are different between DIAP1 and active DRONC versus inactive DRONC.27, 28 The Hid peptide was also able to bind to endogenous DIAP1 when it was mixed with S2 cell lysate (Figure S4-c).
Figure 9
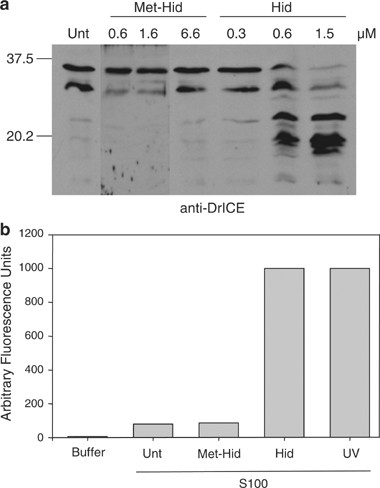
A peptide consisting of the Hid amino terminus (lacking the initiating methionine) is sufficient to induce caspase activation in S100 from untreated S2 cells. (a) S100 from untreated S2 cells was incubated alone (Unt) or with increasing concentrations of Hid or Met-Hid peptide, followed by immunoblotting for DrICE. (b) S100 from untreated S2 cells was incubated alone (Unt) or with Met-Hid peptide (1.6 _μ_M) or Hid peptide (1.5 _μ_M), and caspase activity assay was performed using DEVD-afc as the substrate. UV, S100 from untreated cells was incubated with S100 from S2 cells that had been UV irradiated. Buffer, caspase assay performed using only buffer and DEVD-afc
To determine if inhibiting the interaction between DIAP1 and DRONC was sufficient to induce apoptosis, we transfected the Hid peptide or the control Met-Hid peptide into S2 cells. The transfected Hid peptide formed a complex with endogenous DIAP1 (Figure S4-c, lane 8). Approximately 60% of the S2 cells transfected with the Hid peptide died by apoptosis, as determined by observing cell morphology (data not shown) and by the fact that the cell death was inhibited by z-VAD-fmk, while S2 cells transfected with the Met-Hid peptide did not undergo apoptosis (Figure 10a). Transfection of Hid or Met-Hid peptides into human 293 cells had no affect on cell viability (Figure 10b). These results provide strong evidence that interruption of the DIAP1-DRONC interaction is sufficient to induce caspase activation and apoptosis in S2 cells.
Figure 10
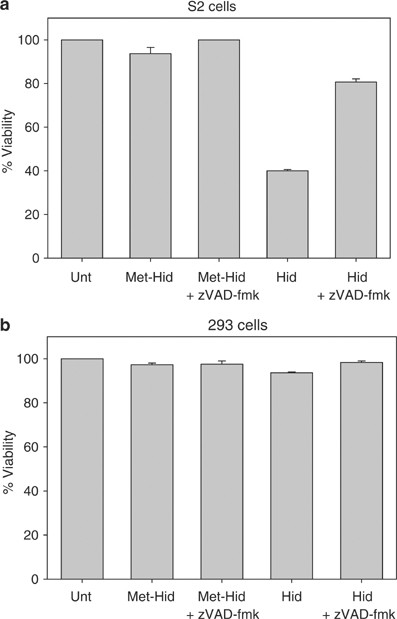
The Hid peptide induces apoptosis when transfected into S2 cells. S2 cells (a) or 293 cells (b) were cotransfected with Hid or Met-Hid peptides and a plasmid expressing eGFP. Cells were treated with zVAD-fmk or carrier control and 48 h later viability was determined. Unt, untreated cells
The data presented so far indicate that mitochondrial factors are not required for caspase activation in S2 cells, but do not rule out a role for these factors in enhancing or accelerating caspase activation. To determine if mitochondria or mitochondrial factors could play a role in enhancing or accelerating caspase activation in vitro, S100 lysate that was isolated from UV-irradiated and zVAD-fmk treated S2 cells was incubated with S2 P10 fraction for 1 h prior to incubation with untreated S2 S100, and at various time points caspase activity was measured. The kinetics of caspase activation in the untreated S100 was not altered by the addition of either intact or lysed mitochondria obtained from either untreated or UV-irradiated S2 cells, indicating that mitochondria or mitochondrial factors do not play a role in caspase activation in this system (Figure 11).
Figure 11
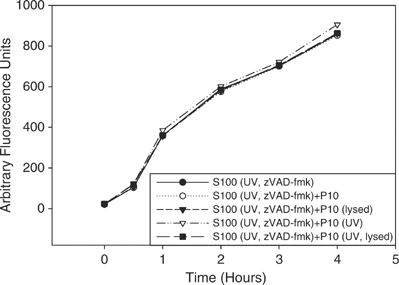
Intact mitochondria or mitochondrial factors do not enhance or accelerate caspase activation in S2 cell lysate. P10 from untreated or UV-irradiated S2 cells were isolated and either lysed or left intact and incubated with S2 S100 that was isolated from UV-irradiated and zVAD-fmk treated S2 cells for 1 h followed by the addition of untreated S2 S100. Caspase activity was measured using Ac-DEVD-afc as substrate
Next, the efficiency of caspase activation by the Hid peptide was compared to full-length Reaper and Hid proteins. Full-length Hid and Reaper proteins that contained either a His tag on the C-terminus of the protein or a glutathione-_S_-transferase (GST) tag on the N-terminus (which would be expected to block the IAP binding motifs) were expressed in bacteria. The Hid peptide and the Reaper and Hid fusion proteins were then compared in their ability to induce caspase activation in vitro. Increasing equimolar concentrations of the Hid peptide, full-length Hid (His- or GST-tagged), Reaper (His- or GST-tagged) or GST were incubated with untreated S2 S100 and caspase activity was measured. At the lowest concentration used, 0.25 _μ_M, caspase activation was greatest for full-length Hid-His, followed by Reaper-His, and finally the Hid peptide (Figure 12a). However, at the next highest concentration (0.6 _μ_M), and at all higher concentrations, the Hid peptide was as efficient as Reaper-His at inducing caspase activation (Figure 12b–f), and at 100 _μ_M the Hid peptide was as efficient as both Reaper-His and Hid-His (Figure 12f).
Figure 12
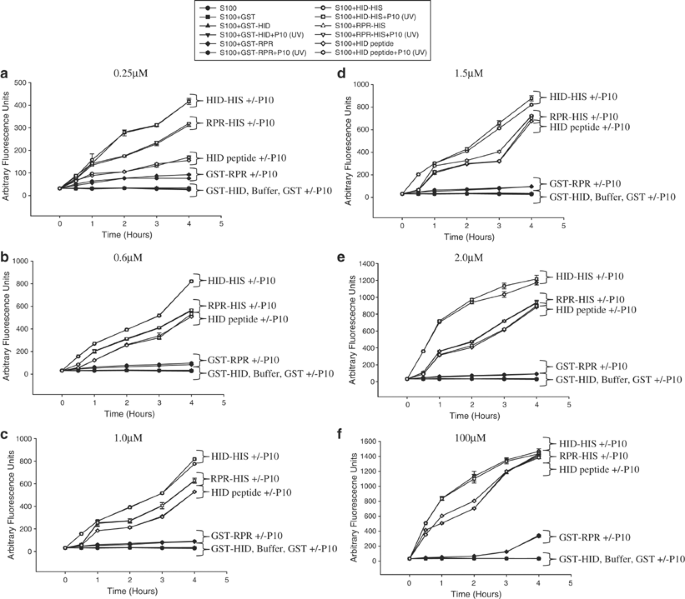
Full-length Reaper and Hid proteins are more efficient at activating caspases compared to the Hid peptide. Recombinant full-length Hid-His, GST-Hid, Reaper-His, GST-Reaper or GST alone were incubated with S2 S100, with or without P10 and at various times after incubation caspase activity was measured using Ac-DEVD-afc as substrate
The observation that full-length Reaper and Hid were more efficient at inducing caspase activation at lower concentrations than the Hid peptide raised the possibility that other domains in Reaper and Hid besides the N-terminal IAP binding domain were contributing to caspase activation, although the IAP binding domain was clearly required for this activity since the GST fusion proteins had little or no activity. Since Reaper has been shown to localize to mitochondria by virtue of its GH3 domain,25 either intact or lysed mitochondria were also added to the reactions to see if they could enhance or accelerate caspase activity. When P10 that was isolated from S2 cells that were UV-irradiated was added, there was no apparent increase or enhancement of caspase activity triggered by Hid-His or Reaper-His (Figure 12). P10 that was isolated from untreated S2 cells also had no effect on caspase activation, and P10 that was subjected to freeze-thawing to lyse the mitochondria after being isolated from either untreated or UV-irradiated S2 cells yielded the same results (data not shown). In addition, the Met-HID peptide showed no caspase-activating ability at any of the concentrations tested (data not shown).
An alternative explanation for why the full-length Reaper and Hid proteins caused caspase activation more efficiently than the Hid peptide is that the full-length proteins caused destabilization of DIAP1 in the S100 lysate. To test this, we incubated the full-length proteins or the Hid peptide with S100 and then analyzed the levels of endogenous DIAP1 in the S100 by immunoblotting. Incubation with either Hid-His or the Reaper-His resulted in decreased levels of DIAP1, while incubation with the Hid peptide did not (Figure S5). Thus, it appears that the full-length proteins were able to induce degradation of DIAP1 in the S100 lysate, which contributed to their ability to cause caspase activation.
Discussion
In mammalian cells, release of cytochrome c is an essential step in mediating caspase activation following stimulation of the intrinsic apoptosis pathway. However, most previous studies have found that cytochrome c is not released during apoptosis in Drosophila S2 cells, and depletion of cytochrome c does not affect apoptosis in Drosophila cells and larvae.21, 22, 23, 24 Previous studies have also shown that mitochondria do not lose their membrane potential during the early stages of apoptosis in Drosophila.22, 24 Consistent with these reports, we also did not observe release of cytochrome c in apoptotic S2 cells following UV irradiation or overexpression of Hid, Reaper, or Grim. When we treated S2 cells with cytochrome c dsRNA to suppress expression of cytochrome c protein and then induced apoptosis, the S2 cells showed no increased resistance to apoptosis and caspase activation was unaffected. It was previously shown that cytochrome c failed to induce caspase activation in insect cell extracts21 and we also found that addition of cytochrome c to S100 did not induce DrICE activation. These data along with ours support the fact that cytochrome c is not required for apoptosis in Drosophila.
Although the requirement for cytochrome c has been examined previously, this is the first report that we are aware of which has examined whether other mitochondrial factors can induce caspase activation in Drosophila cells. In mammals, there is accumulating evidence that the mitochondria are an essential component during intrinsic apoptosis by releasing apoptogenic factors such as cytochrome c,9 AIF,29 Smac/Diablo,30, 31 EndoG,32 or HtrA2/Omi33 from the intermembrane space into the cytoplasm. To determine if there is any involvement of other mitochondrial factors besides cytochrome c for caspase activation in Drosophila, we isolated the mitochondrial and cytosolic fractions from S2 cells that were either normal or apoptotic. None of the P10 fractions were able to induce caspase activation in S100 from normal cells, even when the mitochondria were lysed by freeze–thawing to release their contents. These lysed mitochondria from S2 cells were able to induce caspase activation in S100 from mammalian 293 cells, and cytochrome c appeared to be responsible for this activation. In contrast, cytosolic (S100) fractions isolated from apoptotic S2 cells were able to induce caspase activation in S100 isolated from untreated S2 cells, even when caspase activity was inhibited in the apoptotic S100. This indicated that one or more factors found in the cytosol, which were not caspases, were able to cause caspase activation in normal S100. By using RNAi, we were able to show that Hid, Rpr, and Grim are necessary in an overlapping manner for the caspase-activating activity found in the cytosol of S2 cells.
Using a peptide consisting of amino acids 2–11 of HID, which is able to disrupt DIAP1 binding to DRONC in vitro, we showed that displacement of DIAP1 from DRONC is also sufficient for activation of caspases in S2 S100. This Hid peptide also induced apoptosis when it was introduced into S2 cells. Thus, our results provide the strongest evidence to date that all that is required for caspase activation in S2 cells is interruption of the binding between DIAP1 and DRONC, and that mitochondrial factors do not play a role in Drosophila caspase activation. The possibility remains, however, that mitochondria themselves may play a role in localizing components of the apoptosome in intact cells, as immunofluorescence assays have indicated that a portion of the DRONC and DrICE proteins in Drosophila BG2 cells are localized on the outer surface of mitochondria.24 In agreement with this, we observed DrICE protein in both the S100 and P10 fractions. However, addition of intact mitochondria did not have any stimulatory effect on caspase activation in our in vitro assay, so at least in vitro, there may be sufficient DARK, DRONC, and DrICE in S100 to become activated without the need for mitochondria to bring the proteins together.
Full-length, C-terminally tagged Hid and Reaper proteins caused caspase activation more efficiently than the Hid peptide at the lowest concentration tested, although the Hid peptide was as efficient as Reaper at higher concentrations. The reason why the full-length proteins were more efficient at inducing caspase activation appears to be due to the ability of the full-length proteins to cause DIAP1 destabilization in the S100 lysate, and not to interactions of the full-length proteins with mitochondria or mitochondrial factors. The IAP binding motif was required, since the N-terminally GST-tagged versions had little or no activity in the assay. Removal of the initiating methionine residue from the recombinant proteins, which would be necessary to uncover the IAP binding domain, presumably occurred during expression in the bacteria, as this routinely occurs with bacterially expressed proteins containing alanine in the second position.34 In addition to the IAP binding domain, Reaper has been shown to contain a second motif, called the GH3 domain, that has been reported to be required for DIAP1 degradation, as well as to target Reaper to S2 mitochondria and cause cytochrome c release in mammalian mitochondria.25 The C-terminus of Hid is also capable of mediating mitochondrial localization.25 If these domains of Reaper and Hid were acting by interacting with low level contaminating mitochondrial membranes in the S100, then addition of higher levels of P10 lysate would be expected to cause even higher levels of caspase activation. However, addition of either intact or lysed mitochondria had no effect in our assay, in either the presence or absence of full-length Reaper or Hid. Incubation with Reaper-His or Hid-His did, however, result in decreased levels of DIAP1. This result suggests that proteasome-mediated degradation of DIAP1, which has been shown to be triggered by interaction with Reaper, Hid, or Grim,35, 36 was induced by the addition of Reaper-His or Hid-His to the S100 lysate. This explains why the full-length proteins were more efficient at inducing caspase activation than the Hid peptide.
S100 extract from apoptotic S2 cells was also able to induce caspase activation in 293 whole-cell lysate. This activity appeared to be due to both caspases and Hid, Rpr, and Grim, as inhibition of both of these activities was required to inhibit the caspase activating activity. The observation that apoptotic S2 S100 was not able to induce caspase activation in the absence of mitochondria argues that in 293 S100, Hid, Reaper, and Grim or caspases target the mitochondria and lead to a release of cytochrome c. However, in S2 cells, mitochondria are not required and Hid, Reaper, and Grim appear to target the DIAP1–DRONC complex and remove DIAP1 inhibition of DRONC.
In conclusion, based on these results as well as previous studies, it appears that mitochondrial factors do not play a significant role in Drosophila caspase activation. In S2 cells, DRONC undergoes continuous autoprocessing and this is dependent on DARK.28 DIAP1 binds to DRONC and causes ubiquitination and degradation of autoprocessed DRONC.37 This interaction is required to prevent the cells from undergoing apoptosis. When DIAP1 levels are reduced or when binding of DIAP1 and DRONC is antagonized by Hid, Rpr, or Grim, apoptosis proceeds spontaneously without the need for mitochondrial factors. The possibility remains that in intact Drosophila cells, mitochondria play a role in localizing apoptosome-like complexes, but this does not seem to be required in vitro. It also remains possible that mitochondrial factors play other roles in Drosophila apoptosis, such as inducing DNA cleavage, or have a role in inducing non-caspase mediated cell death. However, our results indicate that factors resident in mitochondria do not play an important role in triggering caspase activation in Drosophila, unlike in mammalian cells. Thus, the reason why Reaper and Hid localize to mitochondria in intact S2 cells may simply be to bring them in close proximity with apoptosome complexes assembled at the mitochondria surface, and not to cause release of mitochondrial factors.
Materials and Methods
Cell culture
Drosophila S2 cells were maintained in Schneider's medium supplemented with 10% fetal bovine serum (FBS). Human 293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 10% Optimem and 1% Glutamax. All cell culture media and supplements were purchased from Invitrogen.
Cell fractionation
Cell fractionation was performed essentially as described previously (Dorstyn et al., 2002). S2 cells (2 × 109 untreated or 1 × 108 treated) or 5 × 106 293 cells were harvested by centrifugation at 400 × g for 5 min. Cell pellets were resuspended in 1 ml of Caspase buffer A (20 mM HEPES·KOH, pH 7.5, 50 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT in 250 mM sucrose supplemented with protease inhibitor cocktail). Cells were homogenized using a Dounce homogenizer by 250 strokes with a tight fitting pestle, and homogenates were centrifuged at 500 × g for 10 min at 4°C. The supernatant was centrifuged at 10 000 × g for 15 min at 4°C and the mitochondrial pellet (P10) was resuspended in 1 ml Caspase Buffer A and stored at −20°C. The supernatant was further centrifuged at 100 000 × g for 30 min at 4°C. The resulting pellet (P100) was resuspended in 500 _μ_l of Caspase Buffer A and stored at −20°C. Glycerol (10%) was added to the resulting supernatant (S100) and the lysate was stored at −80°C.
Recombinant protein and peptide preparation
GST, GST-DIAP1 (N-terminally tagged), DRONC-His6 (C-terminally tagged), DRONC (C318S)-His6 (C-terminally tagged), GST-Reaper (N-terminally tagged), GST-Hid (N-terminally tagged), Hid-His6 (C-terminally tagged), and Reaper-His6 (C-terminally tagged) were expressed in BL21pLysS(DE)3 Eschericia coli (Stratagene). Cultures were grown at room temperature to OD600=0.4, at which time they were induced with 0.1 M IPTG for 1 h, except for Hid-His and GST-Hid, which were grown overnight at 8°C to OD600=0.4, and induced with 0.2 M IPTG. The bacteria were sonicated in lysis buffer A (200 mM Tris-Cl pH 8.0, 0.4 M ammonium sulfate, 10 mM MgCl2, 10% glycerol, and protease inhibitor cocktail (Roche) and purified using either glutathione-sepharose-conjugated beads for GST-DIAP1 or with Talon Metal Affinity Resin (Clontech) for the His6 tagged caspases, according to the manufacturer's instructions. The Met-Hid (MAVPFYLPEGGK-biotin) and Hid (AVPFYLPEGGK-biotin) peptides were synthesized by Sigma-Genosys and were previously described.38
Coimmunoprecipitations
To examine DIAP1 interaction with the Hid peptide, recombinant GST-DIAP1 (0.5 _μ_M) was incubated with increasing concentrations of either the Hid peptide or the Met-Hid peptide at 30°C for 1 h. After incubation, the reaction was added to 50 _μ_l of streptavidin beads and rocked overnight at 4°C. The beads were washed three times with NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM dithiothreitol, 1 mM phenylmethyl sulfonyl fluoride) and bound protein was removed from the streptavidin beads by boiling the samples in Laemmli buffer for 5 min. Proteins were analyzed by immunoblotting using anti-DIAP1 monoclonal antibody at 1 : 100 and anti-mouse IgG-HRP (horseradish peroxidase) antibody at 1 : 4000.
To determine if the Hid peptide could displace DIAP1 from DRONC, 0.5 _μ_g each of GST-DIAP1 and either wild-type DRONC-His6 or catalytically inactive DRONC-His6 (C318S) were mixed together in a total volume of 40 _μ_l of Caspase Buffer A and incubated for 1 h at 30°C. Hid or Met-Hid peptide was then added to the reaction and allowed to incubate for 4 h at 30°C. The reactions were then added to 50 _μ_l of protein G beads (Sigma) that had been preincubated with anti-DRONC antibody (1 : 100) (obtained from Bruce Hay, California Institute of Technology) and rocked overnight at 4°C. The beads were washed three times with Caspase Buffer A and bound protein was removed from the protein G beads by boiling the samples in Laemmli buffer for 5 min. SDS-PAGE and immunoblotting was then performed using anti-DIAP1 monoclonal antibody (obtained from Bruce Hay) at 1 : 100, anti-mouse IgG antibody conjugated with HRP at 1 : 3000 and Supersignal West Pico Chemiluminesent Substrate (Pierce).
To determine if endogenous DIAP1 could bind to Hid peptide, 50 _μ_g of Hid peptide was added to 100 _μ_l of streptavidin-conjugated beads (Sigma) in NP-40 lysis buffer and rocked for 1 h at 4°C to allow the Hid peptide to bind to the beads. The beads were washed two times with NP-40 lysis buffer. 1.4 × 108 S2 cells were harvested in 600 _μ_l of NP-40 lysis buffer and lysed for 30 min at 4°C. After 30 min, the lysates were spun at 21 000 × g at 4°C for 5 min. The lysates were either added to the Hid peptide-bound streptavidin beads and rocked overnight at 4°C or incubated with the Hid peptide for 1 h at 30°C and then added to 100 _μ_l of streptavidin beads and rocked overnight at 4°C. The beads were washed three times with NP-40 lysis buffer and bound protein was removed from the beads by boiling in Laemmli buffer for 5 min. Proteins were analyzed by immunoblotting using anti-DIAP1 monoclonal antibody at 1 : 100 and anti-mouse IgG-HRP antibody at 1 : 4000. The experiment was also performed using S2 S100 instead of whole-cell lysate.
Alternatively, S2 cells (6 × 106 cells per well) were transfected with 3 _μ_g of Hid peptide. After 8 h, cells were harvested in 200 _μ_l/well of NP-40 lysis buffer. Three wells were combined and lysed for 30 min at 4°C. After 30 min the lysates were spun at 21 000 × g at 4°C for 5 min. The supernatant was then added to 50 _μ_l of streptavidin beads and rocked overnight at 4°C. The beads were washed three times with NP-40 lysis buffer and bound protein was removed from the streptavidin beads by boiling the samples in Laemmli buffer for 5 min. Proteins were analyzed by immunoblotting using anti-DIAP1 monoclonal antibody at 1 : 100 and anti-mouse IgG-HRP antibody at 1 : 4000.
RNAi procedure
The RNAi method used was essentially the same as that previously described.37 S2 cells were placed in TC100 medium (Invitrogen) without serum and double-stranded RNA (dsRNA) corresponding in sequence to the entire ORF sequence of the gene being silenced was added directly to the medium at a concentration of 80–120 _μ_g/ml followed by vigorous shaking. The cells were incubated for 5.5 h at 25°C at which time FBS was added to a final concentration of 10%. Cells treated with cytochrome c dsRNA were supplemented with 50 _μ_g/ml uridine and 110 _μ_g/ml pyruvate as in Zimmermann et al.22 To allow time for protein turnover, the cells were incubated for 24–72 h before proceeding with the rest of the experiment.
RT-PCR
S2 cells were treated with dsRNA as described above, and at various times after treatment total RNA was isolated using Trizol reagent (Invitrogen), according to the manufacturer's instructions. Total RNA (3 _μ_g) was used in a reverse transcriptase reaction with a gene-specific primer. From the resulting 50 _μ_l reaction, 2 _μ_l of cDNA was then used as a template in a PCR with nested primers specific for the sequence of interest. In each case, at least one of the primers used for PCR bound to untranslated sequence outside of the open reading frame sequence used to produce dsRNA.
Caspase activity assay
Caspase activity was measured using Ac-DEVD-afc (Enzyme Systems Products) as the substrate. To detect caspase activity in S2 cells that were depleted of cytochrome c, RNAi was performed as described and at 8 h after UV irradiation cells were harvested and spun down at 2000 × g. The cell pellets were resuspended in 100 _μ_l Caspase Buffer A and lysed by four cycles of freeze–thaw and incubated for 1 h at 37°C. 0.2 _μ_M Ac-DEVD-afc was added and the reaction was mixed. The reactions were then analyzed fluorometrically (excitation 405 nm, emission 535 nm) and activity was expressed in relative arbitrary fluorescence units. To detect caspase activity in UV-treated cells, S2 or 293 cells were harvested as described above at various times after UV treatment and incubated with 0.2 _μ_M Ac-DEVD-afc. To detect caspase activity after treatment with the Hid peptide, untreated S100 extract was incubated with Hid or Met-Hid peptide for 4 h at 37°C and then caspase activity was determined as described above. To measure caspase activity in S2 or 293 S100, 25 _μ_l of treated S2 S100 or P10 fractions was added to 150 _μ_l untreated S2 or 293 S100 and incubated at 37°C for 4 h, and then caspase activity was determined.
To examine if mitochondria could enhance or accelerate caspase activity, 25 _μ_l of treated S2 S100 was added to 50 _μ_l of P10 and incubated at 37°C for 1 h followed by the addition of 150 _μ_l untreated S2 S100 and incubated at 37°C for 4 h. Caspase activity was determined at various time points during the 4 h incubation. To determine caspase activity for recombinant RHG proteins, the procedure was identical to the above except that 0.25–100 _μ_M of Hid or Reaper was used instead of treated S2 S100.
Viability assays
To determine the viability with overexpressed Hid, Reaper, or Grim, 106 S2 cells were treated with cytochrome c dsRNA or negative control dsRNA corresponding to the bacterial chloramphenicol acetyl transferase (CAT) gene as described above. Cells were incubated for 72 h and then cotransfected using Cellfectin (Invitrogen) with plasmids expressing enhanced green fluorescent protein (eGFP) (3 _μ_g) and either Hid, Reaper, or Grim (3 _μ_g). These plasmids have been described previously.39, 40 The dsRNA was replaced following transfection (at the time of DNA addition) and 48 h after transfection, the number of viable green fluorescent cells was determined by counting three fields of view under × 400 magnification, and a percent viability was calculated based on the number of eGFP-positive cells in untreated control cells transfected with plasmids encoding eGFP and CAT (set at 100%). For UV viability, the cells were UV irradiated 24 h after transfection by placing the plates on a transilluminator for 10 min. At 24 h after UV treatment, the number of viable, eGFP-positive cells remaining was determined as above and the percent viability was calculated relative to the number of eGFP-positive cells at 1 h after UV treatment. To determine the viability when S2 cells were treated with Hid, Reaper, or Grim dsRNA, 1 × 106 S2 cells were treated with dsRNA as described above and 24 h later the S2 cells were UV irradiated. At 24 h after UV treatment, the percent viability was determined as described above. For viability using the Hid peptide, 1 × 106 S2 cells were cotransfected with eGFP and 3 _μ_g of either Met-Hid peptide or Hid peptide and 48 h after transfection, viability was determined as described above. For experiments using caspase inhibitor, 100 _μ_M zVAD-fmk (Enzyme System Products) was added at the same time as the lipid/DNA mixture and maintained when the media was changed.
Caspase cleavage assay
1 × 108 S2 cells were treated with cytochrome c dsRNA, dronc dsRNA or both as described and UV irradiated. At 4 h after UV treatment, S2 cells were harvested in 1 ml Caspase Buffer A and homogenized, and the P10, P100, and S100 fractions were isolated as described above. In total, 25 _μ_l of either fraction was incubated with 150 _μ_l of untreated S100 prepared from 2 × 109 cells for 4 h at 37°C. To detect caspase cleavage in S100 treated with the Hid peptide, the procedure was similar except that Hid or Met-Hid peptide was incubated with untreated S100. An immunoblot was performed on 15 _μ_l of the sample using anti-DrICE antibody (obtained from Bruce Hay) at a concentration of 1 : 2000 and anti-rabbit IgG-HRP at 1 : 10 000. To examine if cytochrome c addition would induce caspase activation, 150 _μ_l of the S100 was incubated at 37°C for 4 h in the presence of 2 _μ_g of purified cytochrome c (BD Biosciences), 2 mM dATP and 1 mM MgCl2. To examine if mitochondrial factors would induce caspase activation, 150 _μ_l of S100 was incubated with 25 _μ_l of the P10 that was freeze–thawed four times and 15 _μ_l was analyzed by anti-DrICE immunoblot.
Cytochrome c detection
6 × 106 S2 cells were UV irradiated and at various times after UV treatment, the P10 and S100 fractions were isolated and 25 _μ_l was immunoblotted for cytochrome c using 1 : 1000 anti-cytochrome c antibody (BD Biosciences) and 1 : 7000 anti-mouse IgG-HRP. For Hid, Reaper, or Grim, 6 × 106 S2 cells were transfected with plasmids expressing Hid, Reaper, or Grim and 12 h after transfection 10 _μ_l of the P10 and S100 were analyzed for cytochrome c.