Serotonin transporter 5HTTLPR polymorphism and affective disorders: no evidence of association in a large European multicenter study (original) (raw)
Introduction
The serotonin (5-hydroxytryptamine) transporter (5-HTT), responsible for serotonin reuptake into presynaptic neurons, regulates the concentration of serotonin in the synaptic cleft and constitutes a key protein in the complex serotonergic pathway. It has been hypothesized that serotonin is implicated in the pathophysiology of several psychiatric disorders such as affective disorders (AD), autism, obsessive compulsive, and anxiety disorders.1 Moreover, 5-HTT has been reported to have a role in such behaviors as sleep, appetite, aggression, memory, nutrient intake, impulsivity, and personality traits2 (neuroticism, harm avoidance). At a pharmacological level, 5-HTT is the site of binding for tricyclic antidepressants and serotonin reuptake inhibitors – SSRI, thus reducing serotonin reuptake. The 5-HTT gene maps to chromosome 17 at position 17q11.17–q12, and is a strong candidate gene for AD.3
Two 5-HTT polymorphisms have been extensively investigated in AD. The first polymorphism is a variable number of tandem repeats (VNTR) in intron 2, which has been reported to act as a transcriptional regulatory element of the 5-HTT in mice and human.4,5,6 The second VNTR polymorphism is composed of 16 repeat elements, and consists of a 44-bp insertion or deletion (involving repeat elements 6–8), located exactly at the 5′-flanking regulatory region of the serotonin transporter gene on chromosome 17q11.2 (approximately 1 kb upstream of the transcription initiation site), that is, serotonergic transporter-linked polymorphic region (5-HTTLPR). There are two common variants, designated ‘long’ (or ‘L’) and ‘short’ (or ‘S’).7 Studies reported functional repercussions such that the L and S alleles have different transcriptional efficiencies. The L allele has a higher transcriptional activity in vitro and in lymphoblastoid cell lines, compared to SS homozygotes.7,8 Consequently, the rate of removal of serotonin from the synaptic cleft varies according to the distribution of alleles, indicating that the number of repetitive sequences in the 5-HTTLPR polymorphism may have a role in the transcriptional activity. These reports have generated considerable interest, and a very large number of investigations have been conducted on these two 5-HTT polymorphisms in AD. However, conflicting results from association studies do not clarify the role of 5-HTT in phenotypes such as unipolar (UPAD) or bipolar disorders (BPAD) (see the review of Bellivier et al,9 which summarized results of these studies). Additionally, negative linkage studies have also been reported in multiplex families.10,11,12 Lack of statistical power, clinical heterogeneity, genetic heterogeneity, and ethnogeographic stratification as source of bias in association studies may account for the conflicting results. The genetic basis of AD is complex, involving interaction between genetic and environment factors, and most likely involves the interaction of several genes of minor effect. One cannot assume that the presence of an allele is either necessary or sufficient to ‘cause’ the phenotype. The power of association analysis to detect genetic contributions to complex diseases can be greater than linkage studies,13,14 when the sample size gives sufficient power to detect genes of minor effect and when patients and controls are recruited using strict and standardized inclusion criteria, and are well matched for geographical origin. To further clarify the involvement of 5-HTT in AD, we carried out a case–control association study in a large European multicenter sample, using the 5-HTTLPR functional polymorphism. A total of 1932 subjects were selected with DNA and clinical data: 539 unipolar (UPAD), 572 bipolar patients (BPAD), and 821 control subjects (C). To our knowledge, our sample size constitutes one of the largest samples examined for the 5-HTTLPR polymorphism in AD (Table 1).
Table 1 Contribution of each center to the recruitment of patient–control samples
Methods
Subjects
The present sample was recruited within the Biomedical European (BIOMED 2) European Collaborative Study on Molecular Genetics in Affective disorders, Contract No: BMH4-CT-97-2307. This network was established within the framework of the European Commission. The objectives and detailed methodology of the project were described previously.15 A total of eight clinical centers participated in the present study (Edinburgh, Brussels, Sofia, Zagreb, Bonn, Jerusalem, Milan, and Umeå; see Table 1). All subjects were interviewed using standard diagnostic interviews, such as the Schedule for Affective Disorders and Schizophrenia-Lifetime Version (SADS-LA)16 and the Schedule for Clinical Assessment of Neuropsychiatry (SCAN).17 One of the two diagnostic interviews was used for all patients and all controls recruited for the project. The choice of the diagnostic interviews was based on the research experience within individual research teams. Data published by the European Science Foundation (ESF) showed good concordance between the two instruments.18 A total of 1932 subjects were selected with DNA and clinical data: 539 UPAD subjects including 188 males, 349 females, and two missing data, 572 BPAD subjects including 242 males, 328 females, and two missing data, and 821 controls including 363 males, 441 females, and 17 missing data. The age distribution was 53.8 years for UPAD, 49.7 years for BPAD, and 47.3 years for controls. Patients met the diagnosis of BPAD and UPAD, according to RDC, DSM-III-R, and DSM-IV classification systems. To test alternative approaches based on phenotypic traits, we selected clinical subgroups, characterized by a positive family history of AD in first-degree relatives (_n_=94 BPAD; _n_=71 UPAD), antecedents of suicidal attempt (_n_=104 BPAD, _n_=63 UPAD), and presence of psychotic features (delusion or hallucination, _n_=103 BPAD, _n_=17 UPAD). Family data were assessed using the Family History RDC (FH-RDC)19 instrument. When available, family data were also collected from relatives. Positive family history was defined as having at least one first-degree relative affected with BPAD or UPAD. Subjects for whom clinical information to categorize them into those subgroups were not available were not included in these analyses. In the control group, subjects with a positive personal or familial history of psychiatric disorder were excluded. The study was approved by local research ethics committees and informed consent was obtained from patients and controls.
Genotyping
PCR was carried out in a 20–50 _μ_l reaction volume containing 20–100 ng of genomic DNA and 10 pmol of each primer. The primers used in this study were as described by Heils7 et al or Cook20 et al. In all, 200 μ M of dATP, dTTP, and dCTP were used together with 100 μ M dGTP and 100 μ M 7-deaza-dGTP, 1 U of Taq DNA polymerase with the associated buffer containing 1.5 mM Mg2Cl and 5% DMSO. Following heat denaturation of the samples (5 min at 95°C), 35–40 cycles were carried out consisting of 30 s at 94°C, 30 s at 60°C and 30 s at 72°C, followed by a final extension step of 5 min at 72°C. PCR products were resolved on a 3% agarose gel or on a 10% polyacrylamide gel. Bands were visualized by ethidium bromide staining under UV illumination.
Statistical analysis
The Hardy–Weinberg equilibrium (HWE) was tested separately in the three groups (UPAD, BPAD, and controls), by using the exact HW test and the GENEPOP, 3.d. program-updated version of GENEPOP.21 No significant deviation from HWE was observed in any of the three groups. Genotypes’ distribution in controls (LL 33%, LS 47.1%, SS 19.6%) was comparable to that reported by Lesch et al8 in a European population. The empirical statistical power of our two samples (UPAD compared to controls and BPAD compared to controls), based on simulations (odd ratios comprised between 1, 9, and 4 (effect size comprised between 0.16 and 0.3) according to the findings in the literature, and allelic frequencies in controls comprised between 40 and 50%) was more than 90%. It was calculated with program G power.22 Matching in the design was utilized to control for potential confounding factor, such as ethnogeographical origin, a complex nominal variable which represents a wide and indefinable range of environmental and genetic factors difficult to quantify and thus to control by other means. Matching cases and controls in the design were used. Since there were more controls than cases in each center, except Edinburgh, each case was individually matched for ethnogeographical origin to one or several controls within each center (each set was composed of one case and N controls, N varying from a set to another, from 1 to 6). In Edinburgh center, where more cases than controls were ascertained, each control was matched to one or two cases. The number of controls/cases per case/control was allowed to vary from set to set. The same controls were used to be matched with UPAD and BPAD cases. Owing to the matched design, Conditional Logistic Regression for matched sets23 was used to assess the association between dependant variables (diagnosis vs control) and predictors (genotypes/alleles), and to derive odds ratios and 95% confidence intervals after adjustment for potential confounding factors. Estimated coefficients were provided for each of the covariates. We conducted the conditional logistic regression with SPSS, using the coxreg procedure to fit a conditional logit model. The overall _χ_2 (Wald) is provided for each analysis. However, the limitation of matching is the inability to evaluate the effect of matching factor on risk of the outcome. To evaluate the possible modifying effect of center and gender, a stratified analysis was performed (match ignoring) and odds ratios (OR) and confidence intervals (95% CI) were calculated. The Breslow–Day test23 was applied to test the homogeneity of the stratum-specific estimates (center and gender for strata and presence of genotype S–S as exposition). To evaluate the possible confounding effect of gender, the Mantel–Haenszel test24 was applied. Bonferroni correction for multiple tests was applied, when necessary (for four tests: comparisons (patients/controls; patients with family history of AD/controls; patients with suicidal attempt/controls and patients with psychotic features/controls).
Results
Polymorphism HTTLPR and BPAD
The initial focus of our analysis was to detect differences between BPAD and C subjects, in the distribution of genotypes and alleles. No significant differences emerged (_χ_2=0.1, df=2, _P_=0.95 for genotypes and _χ_2=0.07, df=1, _P_=0.8 for alleles distributions – see Figure 1). Subgroup examinations did not show significant differences in genotype and allelic distributions (_χ_2=2.9, df=2, _P_=0.23 and _χ_2=0.98, df=1, _P_=0.32 when considering the group of BPAD with family history of AD compared to controls; _χ_2=2.9, df=2, _P_=0.23 and _χ_2=0.99, df=1, _P_=0.31, the group of BPAD with a history of suicide attempt compared to controls; _χ_2=3.9, df=2, _P_=0.14 and _χ_2=0.43, df=1, _P_=0.5, when considering the group of BPAD with psychotic features compared to controls).
Figure 1
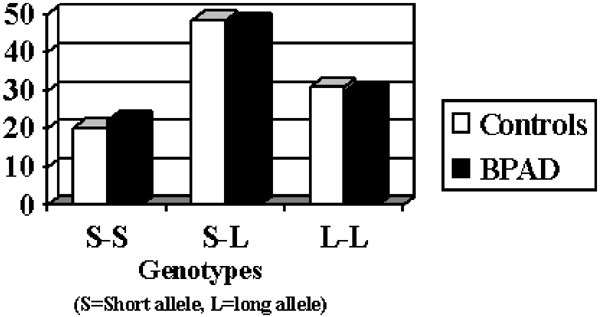
Genotypic distribution (%) between BPAD and controls for 5-HTTLPR.
Stratification tests excluded confounding and modifier effects of gender and center (see Table 2).
Table 2 Stratification tests: Mantel–Haenszel tests (MH) and Breslow–Day test (BD) – dichotomic variable: genotype S–S
Polymorphism HTTLPR and UPAD
No significant association was detected comparing genotype and allele distributions between UPAD patients and control groups (_χ_2=0.62, df=2, _P_=0.73 and _χ_2=0.46, df=1, _P_=0.49, see Figure 2) and subgroups (UPAD with family history of AD/controls _χ_2=7.1, df=2, _P_=0.03 (NS (nonsignificant) after Bonferroni correction for four tests: _P_=0.12; _χ_2=1, df=1, _P_=0.31; UPAD with history of suicidal attempt /controls _χ_2=4.6, df=2, _P_=0.09; _χ_2=0.79, df=1, _P_=0.37; UPAD with psychotic features/controls _χ_2=4.7, df=2, _P_=0.09; _χ_2=0.00, df=1, _P_=0.98).
Figure 2
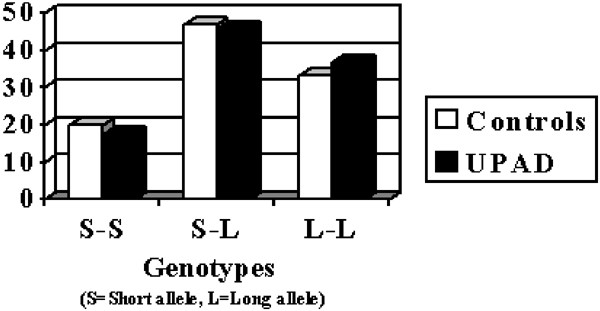
Genotypic distribution (%) between LPAD and controls for 5-HTTLPR.
As for the previous analysis, no confounding or modifier effects emerged for gender and center (see Table 2).
Additional analyses (not shown) performed to test the combined BPAD and UPAD groups, showed no association, when we considered primary diagnosis of AD and phenotypic traits such as suicidal attempt, psychotic features and family history of AD.
Finally, for each group of patients (UPAD and BPAD), we tested the effect of age and age at onset of episode. No age effects emerged on frequencies of genotypes (results not shown).
Discussion
In this multicenter European association study, we collected one of the largest sample sizes of BPAD, UPAD and controls, using strict and standardized inclusion criteria for patients and controls. Our European collaboration has led to efforts to optimize a methodology that reduces some of the most severe limitations of the case–control association approach (such as stratification bias). In particular, we carefully matched patients and controls for geographical origin and used an appropriate statistical method for matched samples. No association was found between the 5-HTTLPR polymorphism and groups of BPAD and UPAD. The very high statistical power and the exclusion of a center effect render our results reliable. For BPAD, our negative results are comparable to most previous independent reports25,26,27,28,29,30,31,32,33,34 Only four studies reported an association between the S allele of 5-HTTLPR and BPAD.35,36,37,38 In the case of family-based studies, three independent studies reported negative findings when testing for preferential transmission of alleles (transmission disequilibrium test) in samples of trios,39,40,41 and two family haplotype analyses showed positive findings.42,43 However, sample sizes suggested that these studies lacked power (substantial risk of statistical Type II error). Our present report also supports the results of most investigations for UPAD, which also failed to detect association with the 5-HTTLPR polymorphism.25,29,33,36,41,44 Only two findings were in favor of an association.35,37 In addition, conflicting association results were reported when considering different phenotypic transonographical behavioral traits and subgroups of AD (seasonal AD, suicidal behavior, anxiety-related traits, personality dimensions, response to antidepressants, see for a review Bellivier et al9). In our studies, subgroup analyses (family history of AD, suicidal attempt and psychotic features) failed to show any association with the 5-HTTLPR polymorphism variants. Owing to our large sample size, it does not seem that our negative findings are due to lack of statistical power. A special effort was made to collect one of the most important databases for this candidate gene in AD. We cannot totally exclude some implication of 5-HTT in some AD phenotypic traits. It is possible that the polymorphism 5-HTTLPR may be close to another adjacent polymorphism involved in AD, without being in linkage disequilibrium. This situation can exist when the population studied is subdivided into small subpopulations carrying mutations descending from different ancestors and present on different haplotypes. When studying the global population, observable associations in subpopulations may become nonsignificant. Another possibility is that the mutation is old enough for the marker and the adjacent susceptibility locus to be in linkage equilibrium again. Data available in a study that compared allelic frequencies of 5-HTTLPR in populations from several geographic regions of the world reported two common alleles, showing that the polymorphism must have arisen early in the evolutionary history of humans.45 However, allele frequencies vary considerably, as a probable consequence of random genetic drift. Above all, as demonstrated by Gelernter's report,45 the large potential for population stratification in studies using this polymorphism may explain some conflicting results, in particular spurious positive findings. Moreover, we cannot exclude the presence of genetic heterogeneity due to population history.
In summary, we conclude that, in our very large European sample, no association was found with primary psychiatric diagnosis of BPAD and UPAD, and some related phenotypic traits. These results are in line with most previous negative reports. The pharmacogenetic study of therapeutic agents involved in the serotonergic pathway remains nevertheless a promising approach for the future.9,46,47
References
- Gelder MG, Lopez-Ibor JJ, Andreasen N (eds) New Oxford Textbook of Psychiatry. Oxford: Oxford University Press; 2000.
Google Scholar - Mazzanti CM, Lappalainen J, Long JC et al: Role of the serotonin transporter promoter polymorphism in anxiety-related traits. Arch Gen Psychiatry 1998; 55: 936–940.
Article CAS Google Scholar - Ramamoorthy S, Bauman AL, Moore KR et al: Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci USA 1993; 90: 2542–2546.
Article CAS Google Scholar - Lesch KP, Balling U, Gross J et al: Organization of the human serotonin transporter gene. J Neural Transm Gen Sect 1994; 95: 157–162.
Article CAS Google Scholar - MacKenzie A, Quinn J : A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc Natl Acad Sci USA 1999; 96: 15251–15255.
Article CAS Google Scholar - Fiskerstrand CE, Lovejoy EA, Quinn JP : An intronic polymorphic domain often associated with susceptibility to affective disorders has allele dependent differential enhancer activity in embryonic stem cells. FEBS Lett 1999; 458: 171–174.
Article CAS Google Scholar - Heils A, Teufel A, Petri S et al: Allelic variation of human serotonin transporter gene expression. J Neurochem 1996; 66: 2621–2624.
Article CAS Google Scholar - Lesch KP, Bengel D, Heils A et al: Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996; 274: 1483.
Article Google Scholar - Bellivier F, Roy I, Leboyer M : Serotonin transporter gene polymorphism and affective disorder-related phenotypes. Curr Opin Psychiatry 2001; 15: 49–58.
Article Google Scholar - Kelsoe JR, Remick RA, Sadovnick AD et al: Genetic linkage study of bipolar disorder and the serotonin transporter. Am J Med Genet 1996; 67: 215–217.
Article CAS Google Scholar - Ewald H, Flint T, Degn B, Mors O, Kruse TA : A functional variant of the serotonin transporter gene in families with bipolar affective disorder. J Affect Disord 1998; 48: 135–144.
Article CAS Google Scholar - Murphy VE, Mynett-Johnson LA, Claffey E et al: Search for bipolar disorder susceptibility loci: the application of a modified genome scan concentrating on gene-rich regions. Am J Med Genet 2000; 96: 728–732.
Article CAS Google Scholar - Risch NJ : Searching for genetic determinants in the new millennium. Nature 2000; 405: 847–856.
Article CAS Google Scholar - Cardon LR, Bell JI : Study designs for complex diseases. Nat Rev Genet 2001; 2: 91–99.
Article CAS Google Scholar - Souery D, Lipp O, Serretti A et al: European Collaborative Project on Affective Disorders: interactions between genetic and psychosocial vulnerability factors. Psychiatr Genet 1998; 8: 197–205.
Article CAS Google Scholar - Endicott J, Spitzer RL : A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry 1978; 35: 837–862.
Article CAS Google Scholar - Wing JK, Babor T, Brugha T et al: SCAN: schedules for clinical assessment in neuropsychiatry. Arch Gen Psychiatry 1990; 47: 589–593.
Article CAS Google Scholar - Farmer AE, Cosyns P, Leboyer M et al: A SCAN–SADS comparison study of psychotic subjects and their first degree relatives. Eur Arch Psychiatry Clin Neurosci 1993; 242: 352–357.
Article CAS Google Scholar - Andreasen NC, Endicott J, Spitzer RL, Winokur G : The family history method using diagnostic criteria. Arch Gen Psychiatry 1977; 34: 1229–1235.
Article CAS Google Scholar - Cook Jr EH, Courchesne R, Lord C et al: Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry 1997; 2: 247–250.
Article Google Scholar - Raymond M, Rousset F : Genepop (ver1. 2): a population genetic software for exact test and ecuniscism. J Hered 1995; 95: 248–249.
Article Google Scholar - Faul F, Erdfelder E, Power G : A Priori, Post-hoc and Compromise Power Analyses for MS-DOS (Computer Program). Bonn, FRG: Bonn University, Department of Psychology; 1992.
Google Scholar - Breslow NE, Day NE : Statistical Methods in Cancer Research. Volume 1 – The Analysis of Case–Control Studies. Lyon: IARC Scientific Publication no. 32; 1980.
Google Scholar - Mantel N, Haenszel W : Statistical aspects of the analysis if data from retrospective studies of disease. J Natl Cancer Inst 1959; 22: 719–748.
CAS PubMed Google Scholar - Kunugi H, Hattori M, Kato T, Tatsumi M, Sakai T, Sasaki T et al: Serotonin transporter gene polymorphisms: ethnic difference and possible association with bipolar affective disorder. Mol Psychiatry 1997; 2: 457–462.
Article CAS Google Scholar - Rees M, Norton N, Jones I et al: Association studies of bipolar disorder at the human serotonin transporter gene (hSERT; 5HTT). Mol Psychiatry 1997; 2: 398–402.
Article CAS Google Scholar - Hoehe MR, Wendel B, Grunewald I et al: Serotonin transporter (5-HTT) gene polymorphisms are not associated with susceptibility to mood disorders. Am J Med Genet 1998; 81: 1–3.
Article CAS Google Scholar - Oruc L, Verheyen GR, Furac I et al: Association analysis of the 5-HT2C receptor and 5-HT transporter genes in bipolar disorder. Am J Med Genet 1997; 74: 504–506.
Article CAS Google Scholar - Ohara K, Nagai M, Tsukamoto T, Tani K, Suzuki Y, Ohara K : Functional polymorphism in the serotonin transporter promoter at the SLC6A4 locus and mood disorders. Biol Psychiatry 1998; 44: 550–554.
Article CAS Google Scholar - Gutierrez B, Arranz MJ, Collier DA et al: Serotonin transporter gene and risk for bipolar affective disorder: an association study in Spanish population. Biol Psychiatry 1998; 43: 843–847.
Article CAS Google Scholar - Vincent JB, Masellis M, Lawrence J et al: Genetic association analysis of serotonin system genes in bipolar affectivedisorder. Am J Psychiatry 1999; 156: 136–138.
Article CAS Google Scholar - Kim DK, Lim SW, Lee S et al: Serotonin transporter gene polymorphism and antidepressant response. Neuroreport 2000; 11: 215–219.
Article CAS Google Scholar - Mellerup E, Bennike B, Bolwig T et al: Platelet serotonin transporters and the transporter gene in control subjects, unipolar patients and bipolar patients. Acta Psychiatr Scand 2001; 103: 229–233.
Article CAS Google Scholar - Minov C, Baghai TC, Schule C et al: Serotonin-2A-receptor and -transporter polymorphisms: lack of association in patients with major depression. Neurosci Lett 2001; 303: 119–122.
Article CAS Google Scholar - Collier DA, Stober G, Li T et al: A novel functional polymorphism within the promoter of the serotonin transporter gene: possible role in susceptibility to affective disorders. Mol Psychiatry 1996; 1: 453–460.
CAS PubMed Google Scholar - Bellivier F, Henry C, Szoke A et al: Serotonin transporter gene polymorphisms in patients with unipolar or bipolar depression. Neurosci Lett 1998; 255: 143–146.
Article CAS Google Scholar - Furlong RA, Ho L, Walsh C et al: Analysis and meta-analysis of two serotonin transporter gene polymorphisms in bipolar and unipolar affective disorders. Am J Med Genet 1998; 81: 58–63.
Article CAS Google Scholar - Ospina-Duque J, Duque C, Carvajal-Carmona L et al: An association study of bipolar mood disorder (type I) with the 5-HTTLPR serotonin transporter polymorphism in a human population isolate from Colombia. Neurosci Lett 2000; 292: 199–202.
Article CAS Google Scholar - Esterling LE, Yoshikawa T, Turner G et al: Serotonin transporter (5-HTT) gene and bipolar affective disorder. Am J Med Genet 1998; 81: 37–40.
Article CAS Google Scholar - Mundo E, Walker M, Tims H, Macciardi F, Kennedy JL : Lack of linkage disequilibrium between serotonin transporter protein gene (SLC6A4) and bipolar disorder. Am J Med Genet 2000; 96: 379–383.
Article CAS Google Scholar - Serretti A, Cristina S, Lilli R et al: Family-based association study of 5-HTTLPR, TPH, MAO-A, and DRD4 polymorphisms in mood disorders. Am J Med Genet 2002; 114: 361–369.
Article Google Scholar - Mynett-Johnson L, Kealey C, Claffey E et al: Multimarker haplotypes within the serotonin transporter gene suggest evidence of an association with bipolar disorder. Am J Med Genet 2000; 96: 845–849.
Article CAS Google Scholar - Kirov G, Rees M, Jones I, MacCandless F, Owen MJ, Craddock N : Bipolar disorder and the serotonin transporter gene: a family-based association study. Psychol Med 1999; 29: 1249–1254.
Article CAS Google Scholar - Nobile M, Begni B, Giorda R et al: Effects of serotonin transporter promoter genotype on platelet serotonin transporter functionality in depressed children and adolescents. J Am Acad Child Adolesc Psychiatry 1999; 38: 1396–1402.
Article CAS Google Scholar - Gelernter J, Cubells JF, Kidd JR, Pakstis AJ, Kidd KK : Population studies of polymorphisms of the serotonin transporter protein gene. Am J Med Genet 1999; 88: 61–66.
Article CAS Google Scholar - Serretti A, Lilli R, Mandelli L, Lorenzi C, Smeraldi E : Serotonin transporter gene associated with lithium prophylaxis in mood disorders. Pharmacogenomics J 2001; 1: 71–77.
Article CAS Google Scholar - Lesch KP, Heils A : Serotonergic gene transcriptional control regions: targets for antidepressant drug development? Int J Neuropsychopharmacol 2000; 3: 67–79.
Article CAS Google Scholar
Acknowledgements
This work was supported by the Association for Mental Health Research (AESM), the European Community Biomed Grant (Grant No CT 92-1217), the National Fund for Scientific Research (NFSR), the Fund for Scientific Research Flanders (FWO) and the Special Research Fund of the University of Antwerp, Belgium.
Author information
Authors and Affiliations
- Department of Psychiatry, University Clinics of Brussels, Erasme Hospital, Free University of Brussels, 808 route de Lennik, Belgium
Julien Mendlewicz, Isabelle Massat & Daniel Souery - Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology (VIB8), University of Antwerp (UIA), Antwerp, Belgium
Jurgen Del-Favero, Lilijana Oruč & Christine Van Broeckhoven - Psychiatry Clinic, University of Sarajevo, Sarajevo, Bosnia and Herzegovina
Lilijana Oruč - Department of Medical Genetics, University of Antwerp (UIA), Antwerp, Belgium
Markus M Nöthen - Department of Psychiatry and the Department of Medical Sciences, University of Edinburgh, UK
Douglas Blackwood & Walter Muir - Department of Neuroscience, University of Edinburgh, Edinburgh, UK
Sharon Battersby - Department of Psychiatry, Biological Psychiatry Laboratory, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
Beny Lerer & Ronen H Segman - Laboratory of Molecular Pathology, University Hospital of Obstetrics, Medical University, Sofia, Bulgaria
Radka Kaneva - Department of Psychiatry, Vita-Salute University, San Raffaele Institute, Milan, Italy
Alessandro Serretti, Roberta Lilli & Christian Lorenzi - Department of Psychiatry, University Hospital ‘REBRO’, University of Zagreb, Zagreb, Croatia
Miro Jakovljevič & Sladana Ivezič - Department of Psychiatry, University of Bonn, Bonn, Germany
Marcella Rietschel - Department of Psychiatry, First Psychiatric Clinic, Alexander University Hospital, Sofia, Bulgaria
Vihra Milanova
Authors
- Julien Mendlewicz
- Isabelle Massat
- Daniel Souery
- Jurgen Del-Favero
- Lilijana Oruč
- Markus M Nöthen
- Douglas Blackwood
- Walter Muir
- Sharon Battersby
- Beny Lerer
- Ronen H Segman
- Radka Kaneva
- Alessandro Serretti
- Roberta Lilli
- Christian Lorenzi
- Miro Jakovljevič
- Sladana Ivezič
- Marcella Rietschel
- Vihra Milanova
- Christine Van Broeckhoven
Corresponding author
Correspondence toJulien Mendlewicz.
Additional information
Supplementary information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)).
Supplementary information
Rights and permissions
About this article
Cite this article
Mendlewicz, J., Massat, I., Souery, D. et al. Serotonin transporter 5HTTLPR polymorphism and affective disorders: no evidence of association in a large European multicenter study.Eur J Hum Genet 12, 377–382 (2004). https://doi.org/10.1038/sj.ejhg.5201149
- Received: 14 March 2003
- Revised: 21 October 2003
- Accepted: 14 November 2003
- Published: 21 January 2004
- Issue Date: 01 May 2004
- DOI: https://doi.org/10.1038/sj.ejhg.5201149