PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism (original) (raw)
Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder and the most frequent cause of parkinsonism. Parkinsonism has a significant genetic component, particularly in cases with an early onset of the disease (before the age of 51 years). To date, three different genes have been reported to be associated with early-onset parkinsonism (EOP): Parkin at PARK21 (OMIM*602544), PINK1 at PARK62 (OMIM*608309), and DJ-1 at PARK73 (OMIM#606324) (for a review see Shen and Cookson4).
For the first two known genes (Parkin and DJ-1), different types of mutations – point mutations, small deletions, and deletions of whole exons – have been described.3, 5, 6, 7, 8, 9 The latter type of mutations is only detectable upon gene dosage analysis.6 For the recently identified PINK1 gene, only point mutations and one four-nucleotide insertion have been reported to date.2, 10, 11, 12, 13, 14
While the frequency of Parkin mutations in EOP patients has been estimated at 10–25%,5 and DJ-1 mutations account for 1%15, 16 to 2%7, 8 of the tested EOP cases, the frequency of mutations in PINK1 still remains to be accurately determined. Although a first study suggested a frequency of 7%, including patients with heterozygous mutations,10 recent findings revealed that the PINK1 mutation rate is much lower in an Irish PD population.13
Factors associated with mutation frequencies in EOP have been studied most extensively in _Parkin_-associated PD and appear to be highly correlated with lower age of onset and positive family history.5 Ethnic background of the patients also affects the mutation rate with frequencies of Parkin mutations ranging from 66% in patients from Japan17 to less than 4% in a US or Serbian EOP population.8, 18 However, such diverse findings may be the result of referral bias at specialty centers, attracting a higher number of patients with a positive family history, suggestive of a genetic cause. Finally, the rate of detected mutations is dependent on the extent of the mutational analysis performed.19
Thus, we investigated, for the first time, the prevalence of mutations in all three known EOP genes in an ethnically homogeneous group of (mostly) sporadic Italian patients by employing a comprehensive screening protocol.
Materials and methods
Patients
After obtaining informed consent, all patients underwent a standardized neurological examination by a movement disorder specialist (GA, PM, PPP). The diagnosis of PD was based on the UK Brain Bank diagnostic criteria (with the exception that positive family history was not regarded an exclusion criterion).20 All patients represent a consecutive series, collected at three Northern Italian Movement Disorder Centers, and did not originate in a geographic isolate. None of them were included in previous Parkin or DJ-1 studies.
Family history was regarded positive if parkinsonism was reported by a first- or second-degree relative. PD was rated using the Unified Parkinson's Disease Rating Scale (UPDRS) III21 and Hoehn and Yahr Scales22 in the off state; major cognitive dysfunction was excluded by applying the Mini-Mental State Examination.23
Mutational analysis
We screened all 12 exons for Parkin mutations by SSCP analysis and gene dosage studies as reported previously.6 To test for DJ-1 mutations, we carried out a dHPLC analysis, and performed gene dosage studies of all six coding DJ-1 exons by quantitative duplex PCR.7 Exons 2–8 of PINK1 were analyzed by dHPLC analysis. Exon 1 was tested in two overlapping parts (1a and 1b) based on the large size (480 bp+exon/intron boundary). Since the PCR for both parts of exon 1 needed additives that can damage the column of the dHPLC system (see On-line Supplementary Information 1), we used SSCP analysis to detect mutations in this exon. For samples that showed a conspicuous band/elution pattern (SSCP/dHPLC) for any exon in any of the three analyzed genes, the entire coding region of the gene in question was sequenced. Quantitative duplex PCR of all PINK1 exons was adapted from that of the Parkin gene6 and was carried out for 55 patients. The amount and quality of the DNA for the remaining 10 samples were not sufficient for quantitative PCR analysis. Exons that carried a heterozygous polymorphism were not included in the quantitative analysis since heterozygous deletions had already been excluded. Primers and assay conditions are available on-line (see On-line Supplementary Information 1 and 2).
Positive controls with known mutations or polymorphisms were available for all exons of Parkin, except for exon 5, for all amplicons of PINK1, and for exon 5 of DJ-1 for the conventional mutational analysis. Regarding the quantitative analysis, the Beta globin gene was coamplified with each individual Parkin, DJ-1, and PINK1 exon, and served as an internal standard. A relative ratio (concentration of each Parkin, DJ-1, or PINK1 exon to concentration of Beta globin) between 0.8 and 1.2 was considered as normal, a heterozygous deletion was expected at the ratio between 0.4 and 0.6, and a heterozygous duplication between 1.3 and 1.7.6 In all, 100 chromosomes of Italian and 100 chromosomes of German control individuals were screened for the presence of the novel mutations and the novel polymorphism. The mean age of the controls at the time of the blood collection was 52.2±14.3 years.
To investigate whether the carriers of recurrent Parkin and PINK1 mutations shared the same haplotype, a set of microsatellite markers in the PARK2 and PARK6 region was used (Table 1). To determine the phase of one of the PINK1 mutation carriers with two base pair substitutions in exon 4, we separated the alleles by cloning the PCR product of exon 4 in the pCRII-Topo vector (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. We selected four insert-bearing colonies, amplified the insert, and sequenced it using vector-specific primers.
Table 1 Haplotype analysis of samples with recurrent mutations in PINK1 (a) and Parkin (b)
Results
Patients
We included 65 unrelated Italian patients (34 women, 31 men). The mean age of onset was 43.2±5.4 (range 25–51) years. Three patients (5%) had a positive family history (an affected parent), compatible with a dominant mode of inheritance. In total, we found five (8%) mutation carriers in our patient group (one woman, four men; average age of onset: 38.2±9.7 (range 25–49) years (Table 2)). None of these patients had a positive family history for parkinsonism, with the possible exception of one case with late-onset bilateral arm tremor in his father (mutational status unknown).
Table 2 Case reports of patients with mutations
Molecular findings
Three patients carried PINK1 mutations, one of which occurred twice (homozygous c.1602_1603insCAA. in exon 8 [p.534_535insQ]; heterozygous c.1602_1603insCAA; heterozygous c.836G>A in exon 4 [p.R279H]; Figure 1). In addition, two patients showed Parkin mutations (c.734A>T in exon 6 [p.K211N] and c.924C>T in exon 7 [p.R275W]; heterozygous c.924C>T).
Figure 1
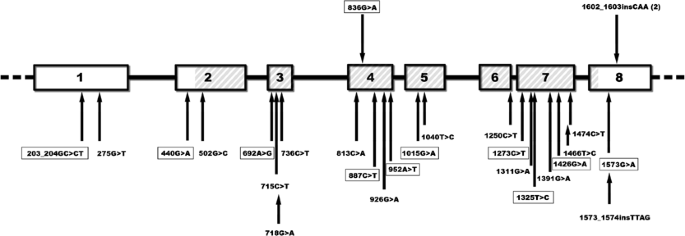
Summary of mutations in the PINK1 gene. A schematic drawing of the eight exons of the PINK1 gene and introns is shown and parts encoding the Ser/Thr protein kinase domain (amino acid 156–509) are hatched. The novel mutations that we found in our sample are indicated above the scheme. The 1602_1603insCAA insertion was found in two patients (once in the homozygous and once in the heterozygous state). The positions of all presently known mutations are marked with black arrows below the scheme of the gene. Mutations detected in the (single) heterozygous state only are boxed.
Genotyping analysis revealed that the two patients with the homozygous and heterozygous PINK1 insertion, respectively, shared at least one allele at each of the six tested microsatellite and the four single-nucleotide polymorphism (SNP) markers at the PARK6 locus. The carrier of the homozygous insertion (Case 1) was heterozygous at five microsatellite markers (Table 1a). The two carriers of the recurrent Parkin mutation (c.924C>T) had alleles in common at four of the five tested markers situated in intronic regions of the Parkin gene, including the rare ‘220 bp’ allele at the marker D6S305 (Table 1b).
None of the patients carried mutations in two different genes, and we did not find any mutations in DJ-1 in our patient sample. Also, there was no gene dosage alteration in any of the genes tested in our patient cohort. The novel PINK1 mutations were not present in 200 control chromosomes.
We detected one as yet undescribed PINK1 polymorphism (c.344A/T), which results in an amino-acid change (p.Q115L) and is present in six out of 100 (6%) screened control individuals. We also found four novel changes of unknown pathogenic significance (−21G/A; IVS1+97A/G; IVS3+38_40delTTT; c.852C/T) (Table 3). Further, 11 known PINK1 polymorphisms were found.
Table 3 Novel changes detected in the PINK1 gene
The patient with the heterozygous c.836G>A mutation in exon 4 of PINK1 also carried the novel c.852C/T substitution (Table 3) that was situated on the same chromosome, 16 bp downstream of the c.836G>A mutation (Figure 2).
Figure 2
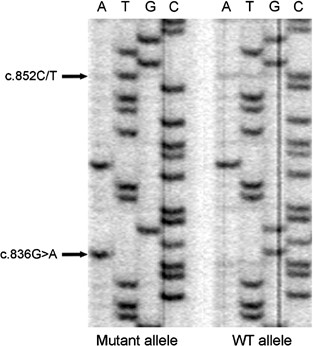
Sequence analysis of clones containing PCR fragments of the mutated (left) and of the wild-type allele (right) of Case 3 demonstrating that both sequence changes (arrows) are localized on the same chromosome.
Discussion
This is the first investigation that comprehensively tested for mutations in all three known genes associated with recessive EOP by employing both conventional methods (SSCP/dHPLC/sequencing) and gene dosage analysis of all coding exons.
Inclusion of PD patients was solely based on the age of onset ≤51 years, regardless of family history, that was negative in the vast majority of our cases. Only one of the five mutation carriers (heterozygous mutation in PINK1) reported that his father had had late-onset, bilateral arm tremor starting at the age of 70 years. No other cases of (possible) parkinsonism were reported in any of the families of our five mutation carriers. In particular, the parents of the carrier of the homozygous PINK1 insertion died at ages 68 and 80 years. Since they had not undergone a detailed neurological examination, it remains unknown whether they had had any (mild) parkinsonian signs. Even if they had been unaffected, this could have been explained by reduced penetrance.
Ages of onset of the heterozygous PINK1 mutation carriers included the youngest (25 years) and one of the oldest ages of onset (49 years) of our patient cohort. It is curious that the heterozygous carrier of the trinucleotide insertion in PINK1 has an earlier age of onset (25 years) than the homozygous carrier of the same mutation (32 years). Thus, in our three PINK1 mutation carriers, we could not confirm a potential correlation of the number of mutations and age of onset, as suggested in a previous study.10 Nevertheless, it is possible that additional genetic and/or environmental factors contribute to the early age of onset. The homozygous carrier first presented with foot dystonia, not supporting the notion that lack of dystonia may be a distinct finding in patients with PINK1 mutations.11 As described previously,10, 11, 12, 13, 14 disease progression was relatively slow in both PINK1 cases with long-term follow-up. Two of the three patients developed depression and autonomic dysfunction. Mild hyperreflexia was present in one case, and sleep benefit in another patient. The two patients with Parkin mutations did not obviously differ clinically from the PINK1 mutation carriers; dystonia was present in one of the Parkin mutation carriers, whereas the other developed depression and sexual dysfunction in addition to the parkinsonian motor signs. All five mutation carriers identified in our study were not clinically different from the remainder of the study cohort. Our findings and the first case descriptions from the literature suggest that patients with Parkin, PINK1, and also DJ-1 mutations may be clinically indistinguishable not only from one another but also from age-matched patients not carrying mutations in any of the three genes.
Both PINK1 mutations (the point mutation and the 3 bp insertion) were novel, whereas both Parkin mutations had been reported before. There was no genealogical evidence that the patients with recurrent mutations were related. However, although chromosomal phase could not be set with certainty, haplotype analysis of the two carriers of the c.1602_1603insCAA mutation would be compatible with a shared allele. Interestingly, however, the homozygous mutation carrier appeared to have two different alleles, suggesting that this mutation has arisen at least twice independently. Alternatively, the insertion may be ancient enough for two recombination events to have occurred delineating the 2.13-cM shared PINK1 and flanking region (Table 1a). A common founder was also conceivable for both carriers of the recurrent Parkin mutation since they shared a rare allele suggestive of a European founder haplotype associated with this mutation.24
Both PINK1 mutations are likely to have a pathogenic role. The arginine to histidine substitution at position 279 in PINK1 is located in the functional Ser/Thr protein kinase domain of the protein. Out of the 24 previously described mutations, 19 are localized in this domain (Figure 1). In addition, the arginine is conserved throughout all species for which data are available in public databases. The role of the silent change (c.852C/T; p.S284S) detected on the same chromosome and only 16 nucleotides away from the c.836G>A (p.R279H) mutation currently remains elusive. It is possible that it is nonpathogenic or that the change leads to abnormalities in splicing. Unfortunately, no RNA was available to test for this possibility. One may speculate that the occurrence of these two changes, so close to one another, may be somehow related.
The p.534_535insQ mutation is located very close to the C-terminus of PINK1 and causes the incorporation of an additional glutamine after two glutamines encoded by another two CAA triplets. This change was not found in any of the control chromosomes. Interestingly, a homozygous mutation in the C-terminus (c.1573_1574insTTAG) has recently been described in a familial EOP case with a similar age of onset at 28 years.12 Another example of a disease-causing, in-frame mutation close to the end of the coding sequence is the GAG deletion in the DYT1 gene that is associated with early-onset torsion dystonia.25
Both detected Parkin mutations (p.L211N and p.R275W) have been previously described in several EOP patients and were thus considered pathogenic.24 The latter point mutation within RING-finger 1 was shown to be the most prevalent mutation among Europeans and probably results from a founder effect.24 It was reported to produce an unusual distribution of the Parkin protein, with large cytoplasmic and nuclear inclusions.26
The rate of PINK1 mutations in EOP is currently largely unknown, but estimated to be almost as high as the frequency of Parkin mutations based on a single study that reported seven PINK1 mutation carriers among 100 EOP patients, including five with heterozygous mutations.10 Taking a different approach, a second investigation evaluated the role of PINK1 mutations in a set of 39 families with autosomal recessively inherited EOP and detected homozygous PINK1 mutations in six out of the eight EOP families that were shown to be linked to the PINK1 chromosomal region.11 One of the most recent studies identified only one heterozygous carrier among 290 unrelated PD patients in Ireland.13 This last result could be explained by the low number of EOP cases (approximately one-third) included in this study, but could also indicate that the PINK1 mutational rate is ethnically dependent. Furthermore, the analysis of a large cohort of patients from North America (consisting of more than 55% EOP patients) revealed a much larger number of mutation carriers.14 Our study design and the results are compatible with those from the recently published study on Italian EOP patients10 and suggest that the frequency of PINK1 mutations may be comparable to that of Parkin mutations in patients with mainly sporadic EOP, at least in some populations.
The prevalence of detected Parkin mutations was low but still in keeping with previous studies on ethnically homogeneous patient samples,8, 18 particularly in sporadic patients. Technical reasons explaining the low mutation rate in the present sample are unlikely, since we employed a comprehensive genetic screening method. This technique was previously demonstrated to detect accurately Parkin mutations in a large number of patients at much higher frequencies.6, 27, 28 With regard to the DJ-1 gene, recent studies demonstrated a mutation frequency between 1 and 2% among EOP patients.7, 8, 15, 16 Only two of the published investigations included testing for gene dosage alterations and, as expected, revealed higher mutation rates (2 versus 1%).7, 8 However, based on those previous investigations, one would only expect one DJ-1 mutation carrier in our 65 study subjects, and the present study confirms the notion that DJ-1 mutations are a rare cause of EOP.
Despite extensive mutational analysis, a considerable percentage of EOP cases have been shown to carry a single heterozygous mutation in the Parkin, DJ-1, and PINK1 gene.10, 14, 16, 29, 30, 31 In addition, preclinical changes have been detected in asymptomatic carriers of heterozygous Parkin mutations using positron emission tomography (PET)32, 33 or transcranial ultrasound.34 Interestingly, haploinsufficiency or a dominant-negative effect have been suggested for heterozygous PINK1 mutations even prior to the identification of the _PARK6_-related gene: asymptomatic, heterozygous carriers of the _PARK6_-associated haplotype revealed a significant reduction in caudate (18)F-dopa uptake in a PET study in comparison with controls.35 However, the potential pathogenic role of heterozygous mutations in ‘recessive genes’ remains a matter of vivid debate. There are several potential ways to address this important issue: (i) comparison of the frequency of single heterozygous mutations among ethnically matched cases and controls as recently described for Parkin36 and PINK1.10, 14 Parkin mutation frequencies were similar between patients and controls,36 whereas PINK1 mutations were much rarer in controls than in patients.10, 14 However, it may be argued that at least some of the controls may not have reached the age of onset yet and have not undergone the same rigorous neurological examination as the cases, so that mild parkinsonian signs may have been present in affected but asymptomatic controls. (ii) (Prospective) examination of heterozygous offspring of homozygous or compound heterozygous mutation carriers, in whom it is much more unlikely that a second mutation may have been overlooked. (iii) Functional studies of the protein. Haploinsufficiency or a dominant effect could be one explanation why we were unable to find a second mutation in two of our PINK1 mutation carriers and in one of the two Parkin mutation carriers (assuming that the second case is compound heterozygous for his Parkin mutations). Alternatively, a second mutation may be localized in an area of the gene that was not screened, such as the promoter or intronic regions.
The finding of PINK1 (and Parkin) mutations in apparently sporadic EOP patients and the relatively high rate of PINK1 mutation carriers (Valente et al10 and present study) suggest that PINK1 mutations are an important cause to be considered in EOP patients, even in cases with a negative family history. Although genotyping a set of tagging SNPs in a cohort of PD patients did not provide evidence for the existence of a common functional variant influencing PD risk in late-onset disease,37 the frequency of PINK1 mutations in ‘idiopathic’, late-onset PD remains to be established. Similarly, frequency and type of PINK1 mutations need to be determined in PD patients of different ethnic background.
References
- Kitada T, Asakawa S, Hattori N et al: Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
Article CAS Google Scholar - Valente EM, Abou-Sleiman PM, Caputo V et al: Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–1160.
Article CAS Google Scholar - Bonifati V, Rizzu P, van Baren MJ et al: Mutation in DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299: 256–259.
Article CAS Google Scholar - Shen J, Cookson MR : Mitochondria and dopamine; new insights into recessive parkinsonism. Neuron 2004; 43: 301–304.
Article CAS Google Scholar - Lücking CB, Dürr A, Bonifati V et al: Association between early-onset Parkinson disease and mutations in the Parkin gene. N Engl J Med 2000; 342: 1560–1567.
Article Google Scholar - Hedrich K, Kann M, Lanthaler AJ et al: The importance of gene dosage studies: mutational analysis of the Parkin gene in early onset parkinsonism. Hum Mol Genet 2001; 10: 1649–1656.
Article CAS Google Scholar - Hedrich K, Djarmati A, Schäfer N et al: DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology 2004; 62: 389–394.
Article CAS Google Scholar - Djarmati A, Hedrich K, Svetel M et al: Detection of Parkin (PARK2) and DJ1 (PARK7) mutations in early-onset Parkinson disease: Parkin mutation frequency depends on ethnic origin of patients. Hum Mutat 2004; 23: 525.
Article Google Scholar - Hering R, Strauss KM, Tao X et al: Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7). Hum Mutat 2004; 24: 321–329.
Article CAS Google Scholar - Valente EM, Salvi S, Ialongo T et al: PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 2004; 56: 336–341.
Article CAS Google Scholar - Hatano Y, Li Y, Sato K et al: Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol 2004; 56: 424–427.
Article CAS Google Scholar - Rohe CF, Montagna P, Breedveld G, Cortelli P, Oostra BA, Bonifati V : Homozygous PINK1 C-terminus mutation causing early-onset parkinsonism. Ann Neurol 2004; 56: 427–431.
Article CAS Google Scholar - Healy DG, Abou-Sleiman PM, Gibson JM et al: PINK1 (PARK6) associated Parkinson disease in Ireland. Neurology 2004; 63: 1486–1488.
Article CAS Google Scholar - Rogaeva E, Johnson J, Lang AE et al: Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol 2004; 61: 1898–1904.
Article Google Scholar - Hague S, Rogaeva E, Hernandez D et al: Early-onset Parkinson disease caused by a compound heterozygous DJ-1 mutation. Ann Neurol 2003; 54: 271–274.
Article CAS Google Scholar - Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW : The role of pathogenic DJ-1 mutations in Parkinson disease. Ann Neurol 2003; 54: 283–286.
Article CAS Google Scholar - Hattori N, Kitada T, Matsumine H et al: Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol 1998; 44: 935–941.
Article CAS Google Scholar - Chen R, Gosavi NS, Langston JW, Chan P : Parkin mutations are rare in patients with young-onset parkinsonism in a US population. Parkinsonism Relat Disord 2003; 9: 309–312.
Article CAS Google Scholar - Klein C, Hedrich K, Wellenbrock C et al: Frequency of Parkin mutations in late-onset Parkinson's disease. Ann Neurol 2003; 54: 415–416.
Article Google Scholar - Gibb WR, Less AJ : A comparison of clinical and pathological features of young- and old-onset Parkinson's disease. Neurology 1988; 38: 1402–1406.
Article CAS Google Scholar - Fahn S, Elton RL, Members of the UPDRS Development Committee: Unified Parkinson's Disease Rating Scale; in Fahn S, Marsden CD, Calne DB, Goldstein M (eds): Recent Developments in Parkinson's Disease, Vol 2. Florham Park, NJ: Macmillan Healthcare Information, 1987, pp 153–163, 293–304.
Google Scholar - Folstein MF, Folstein SE, McHugh PR : ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12: 189–198.
Article CAS Google Scholar - Hoehn MM, Yahr MD : Parkinsonism: onset, progression, and mortality. Neurology 1967; 17: 427–442.
Article CAS Google Scholar - Hedrich K, Eskelson C, Wilmot B et al: Distribution of Parkin mutations: review and case studies. Mov Disord 2004; 19: 1146–1157.
Article Google Scholar - Ozelius LJ, Hawett JW, Page CE et al: The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997; 17: 40–48.
Article CAS Google Scholar - Cookson MR, Lockhart PJ, McLendon C et al: RING finger 1 mutations in Parkin produce altered localization of the protein. Hum Mol Genet 2003; 12: 2957–2965.
Article CAS Google Scholar - Hedrich K, Marder K, Harris J et al: Evaluation of 50 probands with early-onset Parkinson's disease for Parkin mutations. Neurology 2002; 58: 1239–1246.
Article CAS Google Scholar - Kann M, Jacobs H, Mohrmann K et al: Role of Parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 2002; 51: 621–625.
Article CAS Google Scholar - West A, Periquet M, Lincoln S et al: Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 2002; 114: 584–591.
Article Google Scholar - Clark LN, Afridi S, Mejia-Santana H et al: Analysis of an early-onset Parkinson's disease cohort for DJ-1 mutations. Mov Disord 2004; 19: 796–800.
Article Google Scholar - Singleton A : What does PINK1 mean for Parkinson diseases? Neurology 2004; 63: 1350–1351.
Article Google Scholar - Hilker R, Klein C, Ghaemi M et al: Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the Parkin gene. Ann Neurol 2001; 49: 367–376.
Article CAS Google Scholar - Scherfler C, Khan NL, Pavese N et al: Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic Parkin-linked parkinsonism. Brain 2004; 127: 1132–1142.
Article Google Scholar - Walter U, Klein C, Hilker R, Benecke R, Pramstaller PP, Dressler D : Brain parenchyma sonography detects preclinical parkinsonism. Mov Disord 2004; 19: 1445–1449.
Article Google Scholar - Khan NL, Valente EM, Bentivoglio AR et al: Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol 2002; 52: 849–853.
Article Google Scholar - Lincoln SJ, Maraganore DM, Lesnick TG et al: Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord 2003; 18: 1306–1311.
Article Google Scholar - Healy DG, Abou-Sleiman P, Ahmadi KR et al: The gene responsible for PARK6 Parkinson's disease, PINK1, does not influence common forms of parkinsonism. Ann Neurol 2004; 56: 329–335.
Article CAS Google Scholar - Kong A, Gudbjartsson DF, Sainz J et al: A high-resolution recombination map of the human genome. Nat Genet 2002; 31: 241–247.
Article CAS Google Scholar
Acknowledgements
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (KI 1134/2–4), the Bundesministerium für Bildung und Forschung (01GI0201), the Parkinson's Disease Foundation/National Parkinson Foundation, and the Volkswagen Foundation (all to CK), and the Gottlieb Daimler- und Karl Benz-Stiftung (02-23/03) (to AD).
Author information
Author notes
- Christine Klein and Ana Djarmati: These authors contributed equally to this work
Authors and Affiliations
- Department of Neurology, University of Lübeck, Lübeck, Germany
Christine Klein, Ana Djarmati, Katja Hedrich, Nora Schäfer, Norman Kock, Birgitt Schüle, Anja Hiller, Thora Lohnau, Susen Winkler & Karin Wiegers - Department of Human Genetics, University of Lübeck, Lübeck, Germany
Christine Klein, Ana Djarmati, Katja Hedrich, Nora Schäfer, Norman Kock, Birgitt Schüle, Thora Lohnau, Susen Winkler & Karin Wiegers - Faculty of Biology, University of Belgrade, Belgrade, Serbia
Ana Djarmati - Institute of Neurology, University of Bologna, Bologna, Italy
Cesa Scaglione & Paolo Martinelli - Department of Neurology, University of Genova, Genova, Italy
Roberta Marchese & Giovanni Abbruzzese - Department of Medical Genetics, University of Tübingen, Tübingen, Germany
Robert Hering, Peter Bauer & Olaf Riess - Department of Neurology, General Regional Hospital, Bolzano-Bozen, Italy
Peter P Pramstaller - Department of Genetic Medicine, EURAC-Research, Bolzano-Bozen, Italy
Peter P Pramstaller
Authors
- Christine Klein
- Ana Djarmati
- Katja Hedrich
- Nora Schäfer
- Cesa Scaglione
- Roberta Marchese
- Norman Kock
- Birgitt Schüle
- Anja Hiller
- Thora Lohnau
- Susen Winkler
- Karin Wiegers
- Robert Hering
- Peter Bauer
- Olaf Riess
- Giovanni Abbruzzese
- Paolo Martinelli
- Peter P Pramstaller
Corresponding author
Correspondence toChristine Klein.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Klein, C., Djarmati, A., Hedrich, K. et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism.Eur J Hum Genet 13, 1086–1093 (2005). https://doi.org/10.1038/sj.ejhg.5201455
- Received: 21 January 2005
- Revised: 19 May 2005
- Accepted: 19 May 2005
- Published: 22 June 2005
- Issue Date: 01 September 2005
- DOI: https://doi.org/10.1038/sj.ejhg.5201455