Association of the TSHR gene with Graves' disease: the first disease specific locus (original) (raw)
Introduction
Autoimmune thyroid disease (AITD) is a common disorder, encompassing both Graves’ disease (GD) and autoimmune hypothyroidism (AIH) each with a prevalence of around 1%.1 GD manifests through the production of autoantibodies that bind and stimulate the thyroid-stimulating hormone receptor (TSHR) resulting in hyperthyroidism.2 Similarly, in AIH autoimmune targeting of the thyroid leads to tissue destruction and hypothyroidism, although less than 15% of cases are believed to be due to the effect of blocking autoantibodies against the TSHR2 with other thyroid autoantibodies likely to be exerting a primary disease-causing effect.3
Evidence for a genetic predisposition to AITD is well established from familial clustering and twin studies, with sibling-risk estimates of between 5 and 10 for individual diseases.4, 5 Genome scans across extended families and affected sibling pair cohorts have detected linkage to several chromosome regions, but have yet to convincingly replicate a single locus.6, 7 Candidate genes studies have met with greater success, with replicated associations between AITD and HLA on chromosome 6p21,8 CTLA4 on chromosome 2q339 and LYP on chromosome 1p13.10, 11
The well-known biological role of the TSHR and the link between TSHR autoantibodies and disease have made the TSHR an obvious candidate for AITD. While family studies have yet to confirm the linkage between the TSHR region on chromosome 14 and an AITD phenotype,6, 7, 12 several small association studies have examined known amino-acid residue changing polymorphisms within the TSHR13, 14, 15, 16, 17, 18, 19, 20, 21 and produced conflicting results.
In the present study, we sought to determine whether the TSHR is a susceptibility locus for an AITD phenotype. First, by using 40 single nucleotide polymorphisms (SNPs), we defined linkage disequilibrium (LD) ‘blocks’ across the TSHR genomic interval and subsequently selected haplotype tagging SNPs (htSNPs).22, 23 We then conducted a two-stage case–control association study (1059 AITD cases vs 971 controls) using the htSNPs as well as polymorphisms across the TSHR previously analysed by other groups.13, 14, 15, 16, 17, 18, 19, 20, 21 Finally, we replicated the most associated SNP in an independent cohort (1366 AITD cases and 1061 controls).
Materials and methods
Study design
For assay validation and LD analysis, 86 control samples from the UK were genotyped. In order to limit the amount of genotyping, save DNA resources and decrease reagent expenditure, a two-staged approach was used in the initial association study. For Stage 1, 366 AITD cases (183 AIH and 183 GD) and 350 controls were genotyped. Only SNPs showing association, P<0.05, in the Stage 1 analysis were taken forward to Stage 2. For Stage 2, 730 AITD cases (367 AIH and 363 GD) and 621 controls were genotyped. Both index cases and controls were selected randomly from their respective cohorts. The most associated SNP from the combined data set was then analysed in the UK replication cohort.
Subjects (initial study 1)
Unrelated index cases with GD and AIH were taken from 1119 relative pairs with AITD. The relative pairs were collected as part of an international collaboration from eight participating centres in the UK, Continental Europe (Denmark) and Australasia (Australia and New Zealand) all with similar prevalence data for AITD. The study received ethical approval from each institution's local review ethics committee and additionally from the regional multi-centre ethics committees where appropriate. Written informed consent was obtained from every participant. To minimise genetic heterogeneity, only subjects with at least a three-generation history of white Caucasian origin were recruited. All subjects satisfied identical inclusion and diagnostic criteria for the respective disease states and were recruited by the local endocrinologist. Families were recruited if they had either a sibling pair or at least two affected members (including avuncular and grandparent/grandchild pairs, but excluding parent/offspring relationships) affected with AITD.
GD was defined by the presence of documented biochemical hyperthyroidism in combination with either (1) a diffuse goiter on a scan or (2) positive autoantibodies (to TSHR, TG or TPO) or (3) Graves’ ophthalmopathy or (4) confirmation of a lymphocytic infiltrate in thyroid histology. The diagnostic criteria for AIH were defined as documented biochemical hypothyroidism and either (1) positive autoantibodies to TG or TPO or (2) histological confirmation of a lymphocytic infiltrate in a fine needle aspirate or (3) presence of a goiter on clinical examination. Clinical criteria reflected those seen in the general population1, 2, 3 with a mean age at onset within the fourth decade and, for example, 50% of patients with GD having thyroid eye disease (TED) and an AITD F:M ratio of 6:1.
Gender matched controls with no known family history of AITD, of white Caucasian decent, were collected from seven large centres within the UK via general practice.
Subjects (replication study 2)
Unrelated Caucasian patients of UK origin with GD and AIH were recruited from thyroid clinics in the UK, as previously described.24, 25 Briefly, patients were defined as having GD by the presence of biochemical hyperthyroidism together with either the presence of TED or two of the following criteria: diffuse goiter, a significant titre of microsomal (TPO), thyroglobulin (Tg) or TSHR autoantibodies. AIH was diagnosed by the presence of biochemical hypothyroidism together with a significant titre of TPO, Tg or TSHR autoantibodies, with or without a diffuse goiter. Ethnically and gender-matched control subjects with no family history of autoimmune disease were bled at geographically matched sites. All control subjects had normal thyroid function and were negative for thyroid autoantibodies. All patients and subjects gave informed written consent and the local ethics committee approved the project. In total, 1366 AITD cases (307 AIH and 1059 GD) and 1061 controls were recruited.
Genotyping
Prior to genotyping, venous blood samples were taken from all subjects and DNA was prepared centrally (Oxagen Ltd) from whole blood using the Puregene kit (Gentra Systems) according to the manufacturer's instructions. DNA concentration was checked by performing three independent Picogreen assays. Each sample was checked for amplification success using three different sets of primers and products on an agarose gel with 99.9% of DNA samples extracted passing the amplification QC test.
A total of 40 SNPs across a 600 kb genomic DNA interval (NCBI, build 34, chromosome 14) encompassing the TSHR transcript (GenBank NM_000369) were selected for validation from public databases (dbSNP) and previous publications13, 14, 15, 16, 17, 18, 19, 20, 21 (Table S1).
Genotyping for the LD analysis and two-stage experiment was performed using TaqMan® chemistry. All 40 SNPs were ordered as either pre-validated TaqMan® Assays-on-Demand™ or newly designed TaqMan® Assays-by-Design® (Applied Biosystems). Using the RAPIDGene™ automated genotyping system (Oxagen), 2.5 _μ_l of 2 × TaqMan® assay and 2.5 _μ_l of Absolute™ QPCR ROX Mix (ABgene) were added to 10 ng of dried genomic DNA. The TaqMan® reaction was carried out following Applied Biosystems standard protocol. Fluorescent emissions were captured using the ABI Prism® 7900HT Sequence Detection System and converted into genotypes using the ABI Prism® SDS 2.0 software package. Genotype data were electronically linked with phenotype data for each individual.
For the replication study, DNA was prepared in Birmingham UK using Nucleon Bacc II kit from Tepnel Life Sciences PLC, Manchester, UK. rs2268458 was designed and ordered as a PCR-RFLP assay (Sigma Genosys; F-CCAGCAGAGGGAGCACAA, R- TAGAGAATAGAGCAGCAAGACACT). The 415 bp product was amplified from 20 ng of genomic DNA using 35 cycles of PCR (TAQ DNA polymerase, Bioline; dNTPs, Amersham). Postamplification, 5 _μ_l of the 25 _μ_l PCR reaction were digested for 2 h at 37°C using the _Alu_I restriction endonuclease (New England Biolabs). The digested DNA fragments were separated on a 3% agarose gel, visualised using ethidium bromide and converted into genotypes.
Statistical analysis
Pairwise coefficients of LD were estimated using the expectation-maximization (EM) algorithm26 implemented in LDMAX, and the resulting patterns of LD across the TSHR genomic interval were visualised using GOLD 1.1.27 LD ‘blocks’ were defined with a pairwise _r_2 cutoff of >0.25. For each block, haplotype frequencies across all SNPs were estimated using the EM algorithm in SNPHAP v.0.2.1. The minimal set of SNPs which tag all nontag SNPs with a haplotypic _r_2≥0.9 was then selected using the program htsearch, implemented within the statistical package STATA v.1.823 (Table S1).
Significance of single SNP allelic and haplotype association analyses were assessed by a log-likelihood ratio (LLR) test, as described by Clayton.28
Estimation of haplotype frequencies and likelihoods, for both randomised and actual data, was with the software SNPHAP. A PERL script (RASH – randomised association using SNPHAP) was written to automate this procedure and is available on request. In addition to accounting for problems associated with rare haplotypes, the use of an empirical significance level derived by randomising affection status of individuals (rather than of chromosomes) does not require any assumption of Hardy–Weinberg equilibrium in cases or/and controls.28 Finally, for haplotypes, an approximate _Z_-score was computed, to compare the frequency of any individual haplotype in cases and controls. These analysis procedures follow those implemented in the program GENECOUNTING (2002), with the exception that we used SNPHAP to implement haplotype and likelihood estimation. In all, 1 000 000 permutations were used for combined and replication single SNP _P_-value calculations, and 10 000 permutations were used for haplotype and Stage 1 single SNP _P_-value calculations.
Quality of genotypes was assessed with tests of Hardy–Weinberg equilibrium and tests of allelic and haplotypic heterogeneity between the Stages 1 and 2 cohorts. The tests of heterogeneity between the two stages were also conducted by LLR tests with empirical significance levels, here using 10 000 permutations. Odds ratios and corresponding 95% confidence intervals were calculated using GenStat v.6.2 from VSN International Ltd.
Results
SNP validation, LD block determination, and htSNP selection
Three blocks of LD were defined from the pairwise _r_2 data, from 17 SNPs across the TSHR genomic interval (∼250 kb) in 86 individuals. To investigate additional haplotype diversity within the middle block, three further SNPs were validated. To define the ‘boundaries’ of blocks 1 and 3, 17 additional SNPs were validated extending 5′ from block 1 and 3′ from block 3. Finally, to compare current results with those from previous publications, all known amino-acid residue altering polymorphisms were genotpyed. In total, 38 SNPs with minor allele frequencies (MAF) greater than 0.01 were used for LD block structure analysis across the TSHR region (Figures 1 and 2).
Figure 1
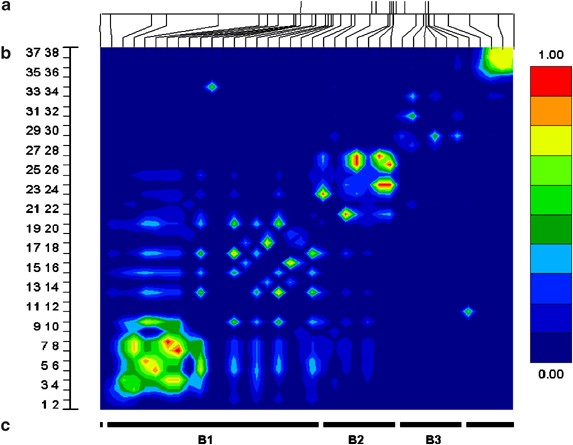
LD analysis across the TSHR genomic region on chromosome 14. (a) Location of the 10 TSHR exons (top) and 38 SNPs (bottom) across the ∼600 kb genomic interval. (b) Graphical interpretation of pairwise _r_2 statistics. (c) LD blocks determined using an _r_2 cutoff of >0.25.
Figure 2
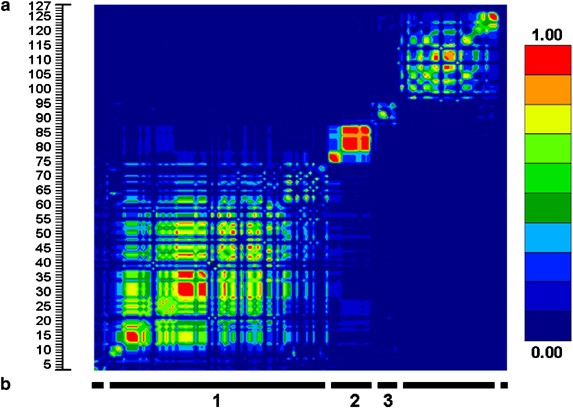
LD analysis across the TSHR genomic region using the Caucasian HAPMAP data. (a) Graphical interpretation of pairwise _r_2 statistics. (b) LD blocks determined using an _r_2 cutoff of >0.25.
Using the National Center of Biotechnology Information (NCBI) build 34 draft of the human genome sequence, the three LD blocks spanning the TSHR were calculated to be roughly 355, 87, and 40 kb, respectively. Block 1 covered the region approximately 280 kb upstream from exon 1 and extended into intron 1. Block 2 started in intron one and terminated at an undefined point between intron 6 and 8. Block 3 covered the remainder of the gene and extended about 10 kb 3′ to exon 10 (Figure 1).
Altogether 18 htSNPs were chosen, nine, four and five SNPs across blocks 1, 2 and 3, respectively. In addition, two extra nontagged SNPs, selected for genotyping prior to defining the 5′ boundary of block 1, were genotyped (Table S1).
Two-stage association study
Stage 1
A total of 20 SNPs were genotyped across 365 AITD cases and 350 controls with an average genotyping call rate of 99.23% (SD 0.78) per marker. Single SNP association (P<0.05) was detected for both GD and AITD, but not for AIH (Table 1). In GD, nine SNPs across blocks 1 and 2 showed single SNP association (P<0.05), including three SNPs with censored _P_-values of 0, due to the use of only 10 000 permutations during analysis (P<1 × 10−4).
Table 1 Stage 1 single SNP association results
The haplotype analysis in Stage 1 gave similar results to the single SNP data, in that GD and AITD showed association with no contribution from AIH (Table 2). While interpretation of the global haplotype results suggested that only block 2 was associated, the _Z_-scores of the individual haplotype results indicated that a single haplotype across blocks 1 and 2 was driving the association (Table 3). When comparing the haplotype data with the single SNP results, this remained the most parsimonious explanation. In fact, all SNPs in blocks 1 and 2 with their minor allele on the associated haplotype showed single SNP association to GD. Only the weak single SNP association with the rare amino-acid-altering polymorphism P52T appeared to be independent to the haplotype.
Table 2 Global haplotype association results (_P_-values)
Table 3 Best individual GD haplotype results (P<1 × 10−4)
As none of the SNPs in block 3 showed association with disease, including rs2288495, which had its minor allele on the associated haplotype, these were not taken forward for further analysis.
Stage 2
The power of the analysis was increased by doubling the number of individuals genotyped and combining the data with Stage 1. An additional 694 AITD cases and 621 controls were genotyped using the 15 SNPs from blocks 1 and 2, with an average genotype call rate of 99.62% (SD 0.69) per marker.
In total, 10 of the 15 SNPs showed association (P<0.05) with GD, three of which were more significant than could be calculated using 1 000 000 permutations (P<1 × 10−6) (Table 4). Investigation into the consistency of the SNP data revealed one deviation from Hardy–Weinberg equilibrium in the combined GD cases (_P_=0.005, rs2284722), one deviation from homogeneity between the two stages of genotyping for the GD cases (_P_=0.012, rs2300523) and one deviation from homogeneity between the various international collection centres for the GD cases (_P_=0.003, rs2300523). No deviations from either Hardy–Weinberg equilibrium or homogeneity were detected in the controls. As in Stage 1, there was no association with AIH.
Table 4 Combined single SNP association results
Stage 2 haplotype association results were also comparable to Stage 1, albeit more significant and with both blocks showing global association with the GD phenotype (Table 2). Analysis of the individual haplotype data indicated that the same haplotype seen in Stage 1 was driving both the global haplotype associations and the majority (9/10) of single SNP associations (Table 3). However, instead of the combined haplotype across blocks 1 and 2 showing the strongest association, analysis of the _Z_-scores suggested that individual haplotypes from each block were driving the combined block association and that the haplotype in block 1 was more associated with GD than the haplotype in block 2. Comparison of the MAFs and single SNP results between blocks 1 and 2 supported this conclusion. SNPs in block 1 with minor alleles on the associated haplotype had MAFs of greater discordance compared to the frequency of the associated haplotype, yet showed stronger single SNP association. Finally, as in Stage 1, the P52T polymorphism was again the only marker to show single SNP association to GD independent of the associated haplotypes.
Replication study
To verify the association between the TSHR and GD detected in the two-stage study, the SNP showing the strongest combined single SNP association, rs2268458 (Table 4), was selected for further investigation in an independent UK AITD cohort. Using alternative genotyping chemistry, rs2268458 was genotyped across 1366 AITD cases and 1061 controls with a genotype call rate of 83.2%. Replicating the previous results, rs2268458 showed association to GD (_P_=2 × 10−6) and no association to AIH (Table 5). Investigation into the consistency of the SNP data between the two studies revealed no heterogeneity (P<0.05) between either the GD cases or the controls. The accuracy of this genotyping method was subsequently validated using TaqMan® chemistry as described above with 100% genotyping replication.
Table 5 Replication results for rs2268458
Discussion
While several studies have previously examined the TSHR for association with AITD, they have met with limited success and consistency, due to a focus on rare amino-acid-altering polymorphisms in relatively small data sets.13, 14, 15, 16, 17, 18, 19, 20, 21 We focused on common SNPs that tagged the limited haplotype diversity in the region, while also including previously studied amino-acid-altering SNPs. Using this approach, we detected highly significant associations to GD but not AIH. P52T was the only previously associated amino-acid-altering SNP to show a weak association to GD in our two-stage experiment, although in the opposite direction to the original study.16 With this discrepancy and the other negative results,13, 15, 17, 18, 19, 21 it would appear that both the initial and our own P52T associations are most likely Type I errors. However, a protective effect of P52T cannot be excluded.
Only one prior AITD genetic study analysed an intronic polymorphism of the TSHR.17 In that study, rs2239610 showed association with an Asian GD cohort. Although this SNP was not analysed in the present study, we did type rs2268458, a SNP in perfect LD (_r_2=1), with rs2239610 and this showed the strongest single SNP association with GD in our combined data set. In our replication cohort, the association between rs2268458 and GD was confirmed, although the odds ratio was smaller than that in our initial study. Bias in the measurement of effect size, normally leading to inflated risk estimates, is common in studies detecting novel association.29 Thus, the odds ratio from our replication study may be a better measure of the effect of this locus on Caucasian GD susceptibility.
We adopted a staged strategy for the study of a candidate gene region for AITD. The initial association study was conducted in two stages to save valuable resources. Analysis of Stage 1, Stage 2 and the combined data sets showed minor differences in allele and genotype distributions confirming the validity of this approach. A common failing of many genetic studies is the inadequate size of data sets.30 This was overcome in the present study with the establishment of an international collaboration in which all recruiting centres worked to identical inclusion criteria and a central laboratory was used for DNA extraction, genotyping and data analysis. Furthermore, the use of an independent replication cohort addressed a number of potential limitations of the initial study. Not only did it confirm the finding of association of the TSHR region with GD, and lack of association with AIH, but it validated our overall strategy and addressed possible sampling problems that could have arisen over the use of unrelated index cases recruited as part of a family-based study and the potential bias of using samples from different geographical locations with control DNA from only the UK.
Although we have provided strong evidence for association of the TSHR region with GD, further studies are required to identify the primary disease-causing DNA variants within this region. Within the associated region spanning LD blocks 1 and 2, two other novel genes of unknown function reside, c14orf61 and c14orf145. Although these novel genes cannot be excluded as harbouring disease-causing polymorphisms, the TSHR is the most attractive candidate for GD susceptibility in the region. The difference observed between GD and AIH in both studies is, however, an important finding. GD and AIH have similar prevalence data1 showing that we had the same power to detect association with both disease states. The fact that the TSHR has been shown to act as the primary autoantigen for GD2 and not AIH3, 31 supports the notion that polymorphism of the TSHR could be a disease-specific locus for GD.
While further studies are required to confirm the exact location of disease-causing polymorphism in this region, intronic TSHR polymorphisms in LD block 1, altering the splicing of exons coding the extracellular domain of the protein, may play a major role in TSHR autoantibody production. The extracellular domain of the THSR is cleaved, forming α and β subunits. Studies have shown that the α subunit is occasionally shed into the plasma.31 Potentially, the derivation of an alternative extracellular domain through alternative splicing could either instigate shedding or convert the wild-type α subunit into a potent autoantigen, both of which may elicit the autoimmune response.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
- Tunbridge WM, Evered DC, Hall R et al: The spectrum of thyroid disease in a community: the Whickham survey. Clin Endocrinol (Oxford) 1977; 7: 481–493.
Article CAS Google Scholar - Weetman AP : Graves’ disease. N Engl J Med 2000; 343: 1236–1248.
Article CAS Google Scholar - Tunbridge WM, Brewis M, French JM et al: Natural history of autoimmune thyroiditis. BMJ 1981; 282: 258–262.
Article CAS Google Scholar - Brix TH, Kyvik KO, Hegedus L : A population-based study of chronic autoimmune hypothyroidism in Danish twins. J Clin Endocrinol Metab 2000; 85: 536–539.
CAS PubMed Google Scholar - Brix TH, Kyvik KO, Christensen K, Hegedus L : Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab 2001; 86: 930–934.
CAS PubMed Google Scholar - Sakai K, Shirasawa S, Ishikawa N et al: Identification of susceptibility loci for autoimmune thyroid disease to 5q31–q33 and Hashimoto's thyroiditis to 8q23–q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet 2001; 10: 1379–1386.
Article CAS Google Scholar - Tomer Y, Ban Y, Concepcion E et al: Common and unique susceptibility loci in Graves and Hashimoto diseases: results of whole-genome screening in a data set of 102 multiplex families. Am J Hum Genet 2003; 73: 736–747.
Article CAS Google Scholar - Simmonds MJ, Howson JM, Heward JM et al: Regression mapping of association between the human leukocyte antigen region and Graves disease. Am J Hum Genet 2005; 76: 157–163.
Article CAS Google Scholar - Ueda H, Howson JM, Esposito L et al: Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003; 423: 506–511.
Article CAS Google Scholar - Smyth D, Cooper JD, Collins JE et al: Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 2004; 53: 3020–3023.
Article CAS Google Scholar - Velaga MR, Wilson V, Jennings CE et al: The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 2004; 89: 5862–5865.
Article CAS Google Scholar - de Roux N, Shields DC, Misrahi M et al: Analysis of the thyrotropin receptor as a candidate gene in familial Graves’ disease. J Clin Endocrinol Metab 1996; 81: 3483–3486.
CAS PubMed Google Scholar - Allahabadia A, Heward JM, Mijovic C et al: Lack of association between polymorphism of the thyrotropin receptor gene and Graves’ disease in United Kingdom and Hong Kong Chinese patients: case control and family-based studies. Thyroid 1998; 8: 777–780.
Article CAS Google Scholar - Ban Y, Greenberg DA, Concepcion ES, Tomer Y : A germline single nucleotide polymorphism at the intracellular domain of the human thyrotropin receptor does not have a major effect on the development of Graves’ disease. Thyroid 2002; 12: 1079–1083.
Article CAS Google Scholar - Chistyakov DA, Savost’anov KV, Turakulov RI et al: Complex association analysis of graves disease using a set of polymorphic markers. Mol Genet Metab 2000; 70: 214–218.
Article CAS Google Scholar - Cuddihy RM, Dutton CM, Bahn RS : A polymorphism in the extracellular domain of the thyrotropin receptor is highly associated with autoimmune thyroid disease in females. Thyroid 1995; 5: 89–95.
Article CAS Google Scholar - Ho SC, Goh SS, Khoo DH : Association of Graves’ disease with intragenic polymorphism of the thyrotropin receptor gene in a cohort of Singapore patients of multi-ethnic origins. Thyroid 2003; 13: 523–528.
Article CAS Google Scholar - Kaczur V, Takacs M, Szalai C et al: Analysis of the genetic variability of the 1st (CCC/ACC, P52T) and the 10th exons (bp 1012–1704) of the TSH receptor gene in Graves’ disease. Eur J Immunogenet 2000; 27: 17–23.
Article CAS Google Scholar - Kotsa KD, Watson PF, Weetman AP : No association between a thyrotropin receptor gene polymorphism and Graves’ disease in the female population. Thyroid 1997; 7: 31–33.
Article CAS Google Scholar - Sale MM, Akamizu T, Howard TD et al: Association of autoimmune thyroid disease with a microsatellite marker for the thyrotropin receptor gene and CTLA-4 in a Japanese population. Proc Assoc Am Physicians 1997; 109: 453–461.
CAS PubMed Google Scholar - Simanainen J, Kinch A, Westermark K et al: Analysis of mutations in exon 1 of the human thyrotropin receptor gene: high frequency of the D36H and P52T polymorphic variants. Thyroid 1999; 9: 7–11.
Article CAS Google Scholar - Daly MJ, Rioux JD, Schaffner SF et al: High-resolution haplotype structure in the human genome. Nat Genet 2001; 29: 229–232.
Article CAS Google Scholar - Johnson GC, Esposito L, Barratt BJ et al: Haplotype tagging for the identification of common disease genes. Nat Genet 2001; 29: 233–237.
Article CAS Google Scholar - Collins JE, Heward JM, Howson JM et al: Common allelic variants of exons 10, 12, and 33 of the thyroglobulin gene are not associated with autoimmune thyroid disease in the United Kingdom. J Clin Endocrinol Metab 2004; 89: 6336–6339.
Article CAS Google Scholar - Heward JM, Allahabadia A, Daykin J et al: Linkage disequilibrium between the human leukocyte antigen class II region of the major histocompatibility complex and Graves’ disease: replication using a population case control and family-based study. J Clin Endocrinol Metab 1998; 83: 3394–3397.
CAS PubMed Google Scholar - Excoffier L, Slatkin M : Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 1995; 12: 921–927.
CAS PubMed Google Scholar - Abecasis GR, Cookson WO : GOLD – graphical overview of linkage disequilibrium. Bioinformatics 2000; 16: 182–183.
Article CAS Google Scholar - Clayton DG : Population association; in Balding DJ, Bishop M, Cannings C (eds): Handbook of Statistical Genetics. Chichester: Wiley, 2003, 2nd edn, pp 939–960.
Google Scholar - Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG : Replication validity of genetic association studies. Nat Genet 2001; 29: 306–309.
Article CAS Google Scholar - Lohmueller KE, Pearce CL, Pike M et al: Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003; 33: 177–182.
Article CAS Google Scholar - Kopp P : The TSH receptor and its role in thyroid disease. Cell Mol Life Sci 2001; 58: 1301–1322.
Article CAS Google Scholar
Acknowledgements
We are grateful to research nurses for assistance with sample collection, Jenny Taylor for initial project management, Jeremy Martin for software development and support, Anna Barrows for DNA extraction and Sarah Cranston for data management. L Hegedus is supported by the Agnes and Knut Moerk Foundation. J Collins is a Diabetes Research and Wellness Foundation PhD student. This work was also supported by Oxagen Ltd and the Wellcome Trust.
Author information
Authors and Affiliations
- Target Discovery, Oxagen Ltd., Abingdon, OX14 4RY, UK
Bryan M Dechairo, Delilah Zabaneh, Gary J Dawson, Angie P Green, Ian Mackay & Alisoun H Carey - Division of Medical Sciences, Institute of Biomedical Research, University of Birmingham, Queen Elizabeth Hospital, Edgbaston, B15 2TH, Birmingham, UK
Joanne Collins, Oliver Brand, Jayne A Franklyn & Stephen C Gough - University Department of Medicine & Therapeutics, Western Infirmary, Glasgow, G11 6NT, UK
John M Connell - Department of Metabolic Medicine, Nuffield Orthopaedic Centre, Oxford, OX3 7LD, UK
John A H Wass - Department of Endocrinology, University Hospital Academisch Medisch Centrum, University of Amsterdam, Amsterdam, PO Box 22700, 1100 DE, The Netherlands
Wilmar M Wiersinga - Department of Endocrinology and Metabolism, Odense University Hospital, Odense C, DK-5000, Denmark
Laszlo Hegedus & Thomas Brix - Kolling Institute and Department of Endocrinology, Royal North Shore Hospital, University of Sydney, Australia
Bruce G Robinson - Department of Endocrinology, Christchurch Hospital, Christchurch, NZ, Private Bag 4710
Penny J Hunt - University of Sheffield Clinical Sciences Centre, Northern General Hospital, S5 7AU, Sheffield, UK
Anthony P Weetman
Authors
- Bryan M Dechairo
You can also search for this author inPubMed Google Scholar - Delilah Zabaneh
You can also search for this author inPubMed Google Scholar - Joanne Collins
You can also search for this author inPubMed Google Scholar - Oliver Brand
You can also search for this author inPubMed Google Scholar - Gary J Dawson
You can also search for this author inPubMed Google Scholar - Angie P Green
You can also search for this author inPubMed Google Scholar - Ian Mackay
You can also search for this author inPubMed Google Scholar - Jayne A Franklyn
You can also search for this author inPubMed Google Scholar - John M Connell
You can also search for this author inPubMed Google Scholar - John A H Wass
You can also search for this author inPubMed Google Scholar - Wilmar M Wiersinga
You can also search for this author inPubMed Google Scholar - Laszlo Hegedus
You can also search for this author inPubMed Google Scholar - Thomas Brix
You can also search for this author inPubMed Google Scholar - Bruce G Robinson
You can also search for this author inPubMed Google Scholar - Penny J Hunt
You can also search for this author inPubMed Google Scholar - Anthony P Weetman
You can also search for this author inPubMed Google Scholar - Alisoun H Carey
You can also search for this author inPubMed Google Scholar - Stephen C Gough
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toStephen C Gough.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Dechairo, B., Zabaneh, D., Collins, J. et al. Association of the TSHR gene with Graves' disease: the first disease specific locus.Eur J Hum Genet 13, 1223–1230 (2005). https://doi.org/10.1038/sj.ejhg.5201485
- Received: 08 March 2005
- Revised: 26 May 2005
- Accepted: 15 July 2005
- Published: 17 August 2005
- Issue Date: 01 November 2005
- DOI: https://doi.org/10.1038/sj.ejhg.5201485