Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53‐dependent cellular senescence (original) (raw)
Abstract
Dysfunctional telomeres induce p53‐dependent cellular senescence and apoptosis, but it is not known which function is more important for tumour suppression in vivo. We used the p53 R172P knock‐in mouse, which is unable to induce apoptosis but retains intact cell‐cycle arrest and cellular senescence pathways, to show that spontaneous tumorigenesis is potently repressed in Terc_−/−_p53 R172P mice. Tumour suppression is accompanied by global induction of p53, p21 and the senescence marker senescence‐associated‐β‐galactosidase. By contrast, cellular senescence was unable to suppress chemically induced skin carcinomas. These results indicate that suppression of spontaneous tumorigenesis by dysfunctional telomeres requires the activation of the p53‐dependent cellular senescence pathway.
Similar content being viewed by others
Introduction
The prevalence of human cancer increases with advancing age, with most of the cancers occurring in older adults. A remarkable characteristic of human carcinomas is their complex cytogenetic profiles, highlighted by the acquisition of multiple chromosomal aberrations (Maser & DePinho, 2002). One mechanism that can produce this unstable genome is linked to the functional status of telomeres. In mammals, telomeres consist of TTAGGG repetitive sequences that terminate in a 3′ single‐stranded G‐rich overhang and are bound by a unique six‐subunit complex that binds to and protects telomeric ends (de Lange, 2005). The synthesis and maintenance of telomeric repeats are mediated by telomerase, and limiting telomerase activity in most somatic cells results in progressive telomere shortening. Critically shortened telomeres are highly recombinogenic, leading to the formation of dicentric chromosomes, anaphase bridges and engagement of the breakage–fusion–bridge (BFB) cycle, and the generation of novel chromosomal variants that could lead to the emergence of a pro‐cancer genome (Artandi et al, 2000; O’Hagan et al, 2002; Maser & DePinho, 2002). Indeed, genomic instability driven by dysfunctional telomeres is associated with the transition from benign lesions to malignant cancer (Chin et al, 2004; Meeker et al, 2004). Together, these results strongly support the idea that dysfunctional telomere‐driven genomic instability promotes the formation of carcinomas.
The p53 tumour suppressor has highly conserved roles in suppressing tumour development, and is functionally abrogated in more than 60% of all human cancers (Lowe et al, 2004). In response to genotoxic stress, p53 initiates cell‐cycle arrest, cellular senescence or apoptosis to suppress tumorigenesis (Evan & Vousden, 2001). Late‐generation _Terc_−/− mice, which are telomerase deficient and have short telomeres, show elevated p53‐dependent apoptosis in highly proliferative cellular compartments, such as the testis and bone marrow (Blasco et al, 1997; Rudolph et al, 1999). In vitro studies showed that telomere shortening also initiates the onset of replicative senescence, a state of permanent cell‐cycle arrest in which cells show characteristic morphological changes and stain for senescence‐associated β‐galactosidase (SA‐β‐gal) activity (Hayflick, 1965; Dimri et al, 1995). Dysfunctional telomeres are sensed as double‐strand DNA breaks, activating the DNA damage response (DDR) checkpoints, including ATM (ataxia telangiectasia mutated) and CHK2 (checkpoint kinase 2) to impinge on the p53–p21 pathway, initiating cellular senescence (Karlseder et al, 1999; d'Adda di Fagagna et al, 2003). In mouse cells, p53 appears to be the only important senescence checkpoint (Campisi, 2005). Owing to its anti‐proliferative effects, cellular senescence is postulated to be a potent tumour suppression mechanism in vivo (Lowe et al, 2004). Indeed, in the presence of a functional p53‐signaling pathway, _Terc_−/− mice having critically shortened telomeres are highly resistant to tumour development (Greenberg et al, 1999; Gonzalez‐Suarez et al, 2000).
To determine the relative contribution of apoptosis compared with cell‐cycle arrest in p53‐mediated tumour suppression by dysfunctional telomeres in vivo, we used a genetic approach on the basis of our previously reported p53 R172P mice (Liu et al, 2004). Mice with two copies of the p53 R172P allele (abbreviated to p53 P/P) are unable to initiate a p53‐dependent apoptosis response when challenged with apoptosis‐inducing stimuli but are competent for p53‐mediated cell‐cycle arrest. This mouse model therefore provides an excellent opportunity to evaluate the in vivo contributions of p53‐dependent apoptosis and cell‐cycle arrest to tumour suppression in the setting of telomere dysfunction.
Results And Discussion
Cellular senescence in p53 P/P MEFs
Although p53 P/P mouse embryo fibroblasts (MEFs) treated with DNA‐damaging reagents induce cell‐cycle arrest, including the activation of p21 (Liu et al, 2004), it is not known whether the p53‐dependent cellular senescence pathway is intact in these cells. We investigated whether telomere dysfunction activates a senescence programme in p53 P/P by infecting these cells with retrovirus expressing myc‐tagged TRF2_Δ_B_Δ_M (telomere repeat‐binding factor 2)—a potent and well‐characterized inducer of telomere dysfunction that rapidly initiates a p53‐dependent senescence response in wild‐type MEFs (Takai et al, 2003). After infection with TRF2_Δ_B_Δ_M retrovirus, cellular proliferation was markedly compromised in primary and p53 P/P MEFs. This was shown by an approxiamte three‐ to sixfold decline in the number of bromodeoxyuridine (BrdU)‐positive cells compared with cells infected with the vector control (Fig 1A), whereas minimal effect on growth was observed in _p53_−/− MEFs. This decline in proliferation capacity correlated with an approximate 2.5‐fold increase in the number of SA‐β‐gal‐positive senescent cells (Fig 1B,C). Consistent with the observation that cells undergoing cellular senescence activate the p53‐dependent p21‐signalling pathway, overexpression of TRF2_Δ_B_Δ_M in p53 P/P MEFs resulted in upregulation of p21 (Fig 1D). In addition, reduction of BrdU incorporation, induction of p21 level and increased number of SA‐β‐gal‐positive senescent cells were also observed when oncogenic H–Ras was overexpressed in p53 P/P MEFs (Fig 1A–C). These results indicate that the cellular senescence pathway is intact in p53 P/P MEFs.
Figure 1
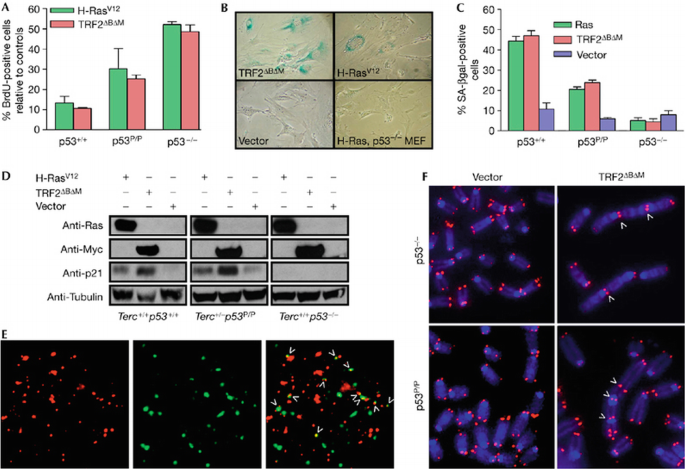
TRF2_Δ_B_Δ_M induces telomere dysfunction and cellular senescence in p53 P/P primary mouse embryonic fibroblasts. (A) BrdU incorporation of p53+/+, p53 P/P and _p53_−/− MEFs at day 4 after infection with H‐RasV12, TRF2ΔBΔM or empty (vector) retrovirus. The percentage of BrdU positive cells relative to controls is indicated. Error bars represent standard error of the mean (s.e.m.). (B) Prominent SA‐β‐gal staining was observed only in p53 P/P but not in _p53_−/− MEFs infected with Myc‐tagged TRF2_Δ_B_Δ_M or H‐RasV12. (C) Quantitation of the percentage of SA‐β‐gal positive cells. Error bars represent s.e.m. (D) Immunoblots of lysates from p53+/+, p53_−/− and p53 P/P MEFs. Cells were infected with either Myc‐_TRF2_Δ_B_Δ_M, H‐RasV12 or vector and proteins were detected with the indicated antibodies. (E) Colocalization of DDR foci to dysfunctional telomeres (arrowheads) in p53 P/P MEFs 48 h after infection with TRF2_Δ_B_Δ_M: telomeres (TRITC, red; left), γ‐H2AX (FITC, green; middle) and merged (right). Arrowheads indicate telomere dysfunction induced foci. (F) End‐to‐end chromosomal fusions were detected after infection with TRF2_Δ_B_Δ_M, but not with vector, in p53 P/P and _p53_−/− MEFs. (E,F) Arrowheads point to sites of fusions. BrdU, bromodeoxyuridine; DDR, DNA damage response; FITC, fluorescein isothiocyanate; MEF, mouse embryonic fibroblast; SA‐β‐gal, senescence‐associated‐β‐galactosidase; TRF2, telomere repeat‐binding factor 2; TRITC, tetramethyl rhodamine isothiocyanate.
Dysfunctional telomeres activate the DDR pathway to initiate p53‐dependent cellular senescence; therefore, we next examined whether a DDR at telomeres is activated in p53 P/P MEFs after infection with the TRF2_Δ_B_Δ_M retrovirus. By using the telomere dysfunction induced foci (TIF) assay (d'Adda di Fagagna et al, 2003; Takai et al, 2003), which monitors the telomeric association of DNA damage proteins, we detected at least four γH2AX‐positive foci in approximately 40% of p53 P/P MEFs, whereas infection with the vector control were only minimally positive for these markers of DNA damage (Fig 1E; supplementary Fig 1A online). This result indicates robust induction of the DDR at telomeres in p53 P/P MEFs. Cytogenetic analyses showed multiple chromosome‐end‐to‐end fusions in these infected cells, characterized by the presence of TTAGGG repeats at the sites of fusion (Fig 1F). Together, these results indicate that the p53‐dependent cellular senescence pathway in p53 P/P MEFs could be induced by dysfunctional telomeres.
Absence of apoptosis in iG1 Terc −/− p53 p/p mice
To genetically dissect the contribution of p53‐dependent apoptosis compared with cellular senescence to tumour suppression in the setting of telomere dysfunction in vivo, we used an intergenerational (iG) mating scheme (supplementary Fig 1B online; Hemann et al, 2001) to generate four experimental cohorts: Terc +/− p53 P/+, Terc +/− p53 P/P, iG1 (first intergenerational mating) Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice, as well as iG1 Terc_−/−_p53+/+, p53 +/− and _p53_−/− controls. Metaphase spreads of primary bone marrow and splenocyte cultures derived from Terc +/− p53 P/+, Terc +/− p53 P/P mice showed minimal structural chromosome abnormalities (Fig 2A,B; data not shown). By contrast, iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P metaphase spreads showed a six‐ to eightfold increase in p–p arm fusions characteristic of chromosomes having dysfunctional telomeres (Fig 2A,B; data not shown).
Figure 2
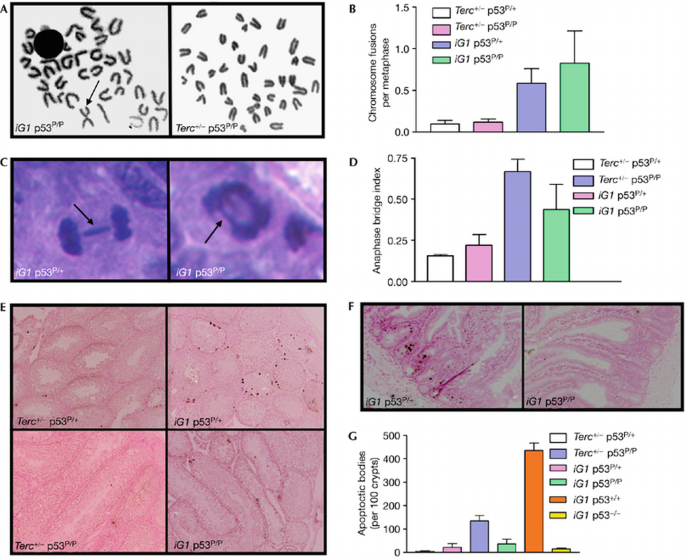
Dysfunctional telomeres do not initiate an apoptotic response in iG1 Terc_−/−_p53 P/P mice. (A) Bone marrow metaphases isolated from iG1 Terc_−/−_p53 P/P mice show chromosome end‐to‐end fusions (arrow), whereas Terc+/−p53 P/P metaphases showed minimal abnormalities. (B) Quantitation of cytogenetic aberrations in bone marrow from mice of the indicated genotypes. Error bars represent s.e.m. (C) Anaphase bridges (indicated by arrows) in intestines of iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice. (D) Quantitation of anaphase bridges in intestines of mice of the indicated genotypes. Error bars represent s.e.m. (E) TUNEL staining of testes of the indicated genotypes. (F) TUNEL staining of intestines of the indicated genotypes. (G) Quantitation of the number of apoptotic cells present in intestinal crypts of the indicated genotypes. Error bars represent s.e.m. iG1, first intergenerational mating; TUNEL, terminal deoxynucleotidyl transferase‐mediated dUTP nick end labelling.
We examined next the intestinal epithelium, a highly quantitative tissue compartment to assess in situ telomere dynamics in vivo. Compared with Terc +/− p53 P/+ and Terc +/− p53 P/P intestines, a two‐ to threefold increase in telomere dysfunction, as measured by an increased number of anaphase bridges in epithelial crypts, was detected in iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P intestines (Fig 2C,D). In iG1 Terc_−/−_p53 P/+ mice, this increase in anaphase bridge index was paralleled by a corresponding increase in TUNEL‐positive apoptotic bodies in both the testes (Fig 2E) and in intestinal crypt compartments (Fig 2F). However, apoptosis was markedly attenuated in iG1 Terc_−/−_p53 P/+ mice compared with iG1 Terc_−/−_p53+/+ mice. It is important to note that apoptosis was absent in both the testis and the intestinal epithelium of iG1 Terc_−/−_p53 P/P mice having a comparable number of anaphase bridges (Fig 2E,F). These results indicate that dysfunctional telomeres do not appreciably activate p53‐dependent apoptosis in tissue compartments of iG1 Terc_−/−_p53 P/P mice.
Tumour suppression in Terc − /− p53 P/P mice
Dysfunctional telomeres drive tumorigenesis under the conditions of p53 deficiency (Chin et al, 1999; Artandi et al, 2000). To determine whether p53‐dependent apoptosis is required to suppress spontaneous tumorigenesis, we monitored tumour development in our mouse cohorts over a 28‐month period. Consistent with a previous report that p53 P/P mice are tumour resistant (Liu et al, 2004), Terc +/− p53 P/+ and Terc +/− p53 P/P controls showed a delay in tumour onset compared with p53 +/− and _p53_−/− mice, with a median tumour latency of 22.6 months for Terc +/− p53 P/+ mice and 9 months for Terc +/− p53 P/P mice (Fig 3A,C). The presence of dysfunctional telomeres was associated with a near complete suppression of tumour formation in both iG1 Terc_−/−_p53 P/+ (0 out of 23 mice with tumours, _P_=0.042) and iG1 Terc_−/−_p53 P/P mice (1 out of 9 mice with tumours, _P_=0.041), which is comparable to the degree of tumour suppression observed in iG1 Terc_−/−_p53+/+ controls (2 out of 11 mice with tumours, _P_=0.01; Fig 3A,C). By sharp contrast, 14 out of 16 _iG1 Terc_−/−_p53_−/− and 6 out of 14 iG1 Terc_−/−_p53 +/− mice readily succumbed to tumours (Fig 3C). Together, these results strongly indicate that p53‐dependent apoptosis is dispensable in mediating telomere‐dependent spontaneous tumour suppression in vivo. Despite this profound cancer resistance, the overall lifespan of iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice was not statistically different from those of their iG1 Terc_−/−_p53+/+ and iG1 Terc_−/−_p53 +/− counterparts (Fig 3B,D).
Figure 3
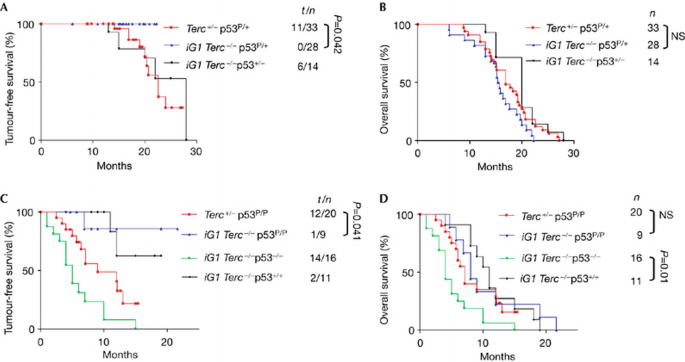
Kaplan–Meier analysis of mouse cohorts. Tumour incidence (A,C) and overall survival (B,D) of mice of the indicated genotypes are shown. The number of tumours identified (t), the total number of mice (n) and the _P_‐values (calculated using the log‐rank test) are indicated. Note the near‐complete tumour suppression in iG1 p53 P/+, iG1 p53 P/P and iG1 p53+/+ mice. Deletion of p53 reduced the lifespan of iG1 mice owing to increased tumour formation. iG1, first intergenerational mating; NS, statistically not significant.
Dysfunctional telomeres induce senescence in vivo
Our observation that dysfunctional telomeres activate p53‐dependent cellular senescence in p53 P/P MEFs prompted us to investigate whether tumour suppression in iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice is associated with activation of cellular senescence in vivo. Immunohistochemical staining with antibodies against p21 and p53 showed abundant positive cells in intestinal epithelium from iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice (Fig 4A,B). By contrast, minimal staining was observed for Terc +/− p53 P/+ and Terc +/− p53 P/P intestines. SA‐β‐gal staining of cryosections from iG1 Terc_−/−_p53 P/+ tissues, including lung, kidney, liver, spleen and small intestine, showed numerous senescent cells, whereas iG1 p53+/+, iG1 p53_−/−_and Terc +/− p53 P/P tissues contained only few positive cells (Fig 4C; supplementary Fig 1C online). These results indicate that the p53‐p21‐dependent cellular senescence pathway is potently activated throughout diverse cellular compartments of iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice, and might be responsible for the observed tumour suppression in these animals. Attesting to this robust tumour suppression, B‐cell lymphoma was detected in only one out of nine iG1 Terc_−/−_p53 P/P mice. Cytogenetic analysis showed that this lesion has multiple chromosomal fusions and telomere‐free chromosome ends (Fig 4D), suggesting activation of the BFB cycle and selection of a pro‐cancer genome that could readily bypass senescence‐mediated cell‐cycle arrest and progress to cancer (Artandi et al, 2000; O’Hagan et al, 2002).
Figure 4
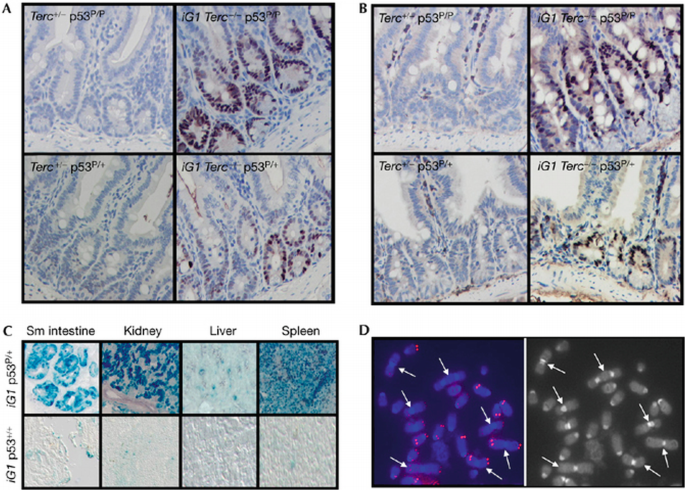
Telomere dysfunction induces cellular senescence in vivo. (A) Immunostaining with p53 antibody detected staining in intestinal crypts of iG1 iG1 Terc_−/−_p53 P/P and Terc_−/−_p53 P/+ mice, but not in Terc+/−p53 P/P and Terc+/−p53 P/+ mice. (B) Immunostaining with p21 antibody detected staining in intestinal crypts of mice of the indicated genotypes. (C) Robust SA‐β‐gal activity detected in tissues from iG1 Terc_−/−_p53 P/+ mice (top) but not in tissues from iG1 Terc_−/−_p53+/+ mice (bottom). (D) End‐to‐end chromosomal fusions (indicated by arrows) and telomere signal‐free ends in an iG1 Terc_−/−_p53 P/P lymphoma. iG1, first intergenerational mating.
Although cellular senescence potently suppressed spontaneous tumorigenesis, inappropriate engagement of the senescence programme is likely to effect organismal survival negatively, as deletion of p21 in mice with dysfunctional telomeres prolonged organismal lifespan and rescued defects observed in highly proliferative stem‐cell compartments, including those of the intestine and haematopoietic system (Choudhury et al, 2006). Although extensive analyses at necropsy failed to reveal a definitive cause of death, several iG1 Terc_−/−_p53 P/P mice showed prominent features of premature ageing, including hair graying and kyphosis, which might be attributable to premature senescence of stem cell reserves (W.C.‐B. and S.C., unpublished data).
Senescence does not suppress DMBA‐induced skin cancer
The classic chemically induced skin carcinogenesis model associated with Ras‐activating mutations shows that dysfunctional telomeres potently suppress skin tumorigenesis in the presence of wild‐type p53 (Fig 5A; Gonzalez‐Suarez et al, 2000). To assess the role of cellular senescence in mediating skin tumour suppression in the setting of telomere dysfunction, we treated mouse cohorts with the tumour initiator DMBA (7,12‐Dimethylbenz[a]anthracene) and then TPA (12‐O‐tetradecanoyl‐phorbol 13‐acetate) twice per week for 20 weeks to promote the development of papillomas. After 11 weeks of TPA treatment, skin lesions appeared in iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice, whereas Terc +/− p53 P/+, Terc +/− p53 P/P mice developed papillomas after 15 weeks (Fig 5A). iG1 Terc_−/−_p53 P/P skin lesions showed an approximately twofold increase in anaphase bridge index over Terc +/− p53 P/P lesions, suggesting the presence of dysfunctional telomeres (Fig 5B). Surprisingly, the number and size of papillomas were greatest in carcinogen‐treated iG1 Terc_−/−_p53 P/P mice: five out of seven treated mice in this cohort developed papillomas that exceeded 10 mm in size, and all of these lesions were premalignant, highly proliferative squamous papillomas/keratoacanthomas with elevated staining for Ki‐67 (Fig 5C). Four out of five mice showed multiple invasive squamous‐cell carcinomas (SCC) with reduced Ki‐67 staining (Fig 5C), presumably owing to the presence of dysfunctional telomeres activating BFB cycles that interfere with the completion of mitosis (Fig 5B). By contrast, only 1 out of 5 Terc +/− p53 P/P mice developed invasive SCC. Robust staining for p53 and p21 was observed in all premalignant lesions as well as invasive SCC (Fig 5C). These results indicate that induction of p53–p21‐mediated cell‐cycle arrest/cellular senescence by dysfunctional telomeres was insufficient to suppress carcinogen‐induced skin tumorigenesis. It is also likely that p53‐mediated apoptosis is required to fully suppress these malignancies (Zhao et al, 2006).
Figure 5
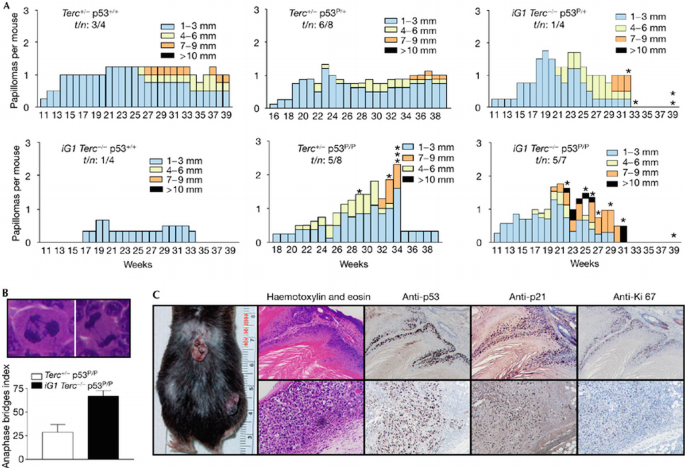
Skin papillomas in mice of the indicated genotypes. (A) Total number of papillomas of the indicated sizes plotted against the number of weeks after carcinogen treatment. The genotypes of treated mice are indicated. Asterisks indicate the week when a mouse died. n: total number of treated mice; t: number of mice with tumours. (B) Increased anaphase bridge index in iG1 Terc_−/−_p53 P/P papillomas. (C) Immunohistochemistry of carcinogen‐treated iG1 Terc_−/−_p53 P/P mouse (left) show high p21, p53 and Ki‐67 positivity in the nuclei of keratinocytes. Upper panel: squamous papilloma. Lower panel: invasive SCC. iG1, first intergenerational mating; SCC; squamous‐cell carcinomas.
Telomere‐induced cellular senescence is postulated to be important in ageing and cancer. However, this process is typically studied in cultured cells, and how it contributes to tumour suppression in vivo has been poorly defined. By using a p53 mutant mouse that is unable to initiate a p53‐dependent apoptosis response when challenged with genotoxic stress but is competent to execute p53‐mediated cellular senescence, our study provides the first direct evidence, to our knowledge, that dysfunctional telomeres induce p53‐mediated cellular senescence to suppress spontaneous tumorigenesis in vivo. In this system, p53‐dependent apoptosis appears dispensable for spontaneous tumour suppression. Dysfunctional telomere‐induced senescence was accompanied by robust increases in p53, p21 and SA‐β‐gal activity in all examined tissue compartments, suggesting that a DDR is activated by dysfunctional telomeres in vivo. Recent observations indicate that the DDR is activated at the earliest stages in many human carcinomas (Bartkova et al, 2005; Gorgoulis et al, 2005). Our results predict that activation of an intact DDR pathway by dysfunctional telomeres in premalignant lesions would result in senescence, suppressing further tumour progression. However, the increased genome instability in nascent tumour cells that stochastically inactivate components of the DDR pathways would promote tumour progression.
Cellular senescence can also be induced by oncogenes, and this oncogene‐induced senescence has been shown to be a potent tumour suppressor mechanism in vivo. Senescent markers were prominent in premalignant lesions but undetectable in advanced cancers (Braig et al, 2005; Chen et al, 2005; Collado et al, 2005; Michaloglou et al, 2005). Recent data indicate that oncogenes induce a robust DDR that initiates the onset of cellular senescence (Bartkova et al, 2006; Di Micco et al, 2006). Together with data presented here, it appears that activation of cellular senescence by either dysfunctional telomeres or oncogenes potently inhibits tumour progression in vivo; thereby placing cellular senescence on an equal footing with apoptosis in mediating tumour suppression.
Methods
Production of mTerc −/− p53P/P mice and initiation of skin carcinogenesis. To reduce the number of generations required to achieve critical telomere shortening, we carried out a cross between Terc+/+p53 P/P mice and generation five (G5) Terc_−/−_p53+/+ mice to generate our starting Terc+/−p53_P/+ animals. Terc+/−_p53_P/+ mice were intercrossed in a cousin‐mating scheme to generate iG1 Terc_−/−_p53 P/+ and iG1 Terc_−/−_p53 P/P mice (Hemann et al, 2001). In addition, Terc+/−_p53 P/+ and Terc+/−p53 P/P mice were generated as cohorts having wild‐type telomere lengths. Control iG1 Terc_−/−_p53+/+, iG1 Terc_−/−_p53+/− and _iG1 Terc_−/−_p53_−/− mice were generated by using a similar mating scheme. Carcinogen‐induced skin carcinogenesis was carried out as described by Gonzalez‐Suarez et al (2000). Skin lesions were fixed in formalin, stained with haematoxylin and eosin, and examined without knowledge of the genotype.
Detailed experimental methods can be found at supplementary information online.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
References
- ArtandiSE, ChangS, LeeSL, AlsonS, GottliebGJ, ChinL, DePinhoRA (2000) Telomere dysfunction promotes non‐reciprocal translocations and epithelial cancers in mice. Nature406: 641–645
Google Scholar - BartkovaJ_et al_ (2005) DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature434: 864–870
Google Scholar - BartkovaJ_et al_ (2006) Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature2444: 633–637
Google Scholar - BlascoMA, LeeHW, HandeMP, SamperE, LansdorpPM, DePinhoRA, GreiderCW (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell91: 25–34
Google Scholar - BraigM, LeeS, LoddenkemperC, RudolphC, PetersAH, SchlegelbergerB, SteinH, DorkenB, JenuweinT, SchmittCA (2005) Oncogene‐induced senescence as an initial barrier in lymphoma development. Nature436: 660–665
Google Scholar - CampisiJ (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell120: 513–522
Google Scholar - ChenZ_et al_ (2005) Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature436: 725–730
Google Scholar - ChinK_et al_ (2004) In situ analyses of genome instability in breast cancer. Nat Genet36: 984–988
Google Scholar - ChinL, ArtandiSE, ShenQ, TamA, LeeSL, GottliebGJ, GreiderCW, DePinhoRA (1999) p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell97: 527–538
Google Scholar - ChoudhuryR_et al_ (2006) Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet39: 99–105
Google Scholar - ColladoM_et al_ (2005) Tumour biology: senescence in premalignant tumours. Nature436: 642
Google Scholar - d'Adda di FagagnaF, ReaperPM, Clay‐FarraceL, FieglerH, CarrP, Von ZglinickiT, SaretzkiG, CarterNP, JacksonSP (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature426: 194–198
Google Scholar - de LangeT (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev19: 2100–2110
Google Scholar - Di MiccoR_et al_ (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature444: 638–642
Google Scholar - DimriGP_et al_ (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA92: 9363–9367
Google Scholar - EvanGI, VousdenKH (2001) Proliferation, cell cycle and apoptosis in cancer. Nature411: 342–348
Google Scholar - Gonzalez‐SuarezE, SamperE, FloresJM, BlascoMA (2000) Telomerase‐deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet26: 114–117
Google Scholar - GorgoulisV_et al_ (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature434: 907–913
Google Scholar - GreenbergRA, ChinL, FeminoA, LeeKH, GottliebGJ, SingerRH, GreiderCW, DePinhoRA (1999) Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer‐prone mouse. Cell97: 515–525
Google Scholar - HayflickL (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res37: 614–636
Google Scholar - HemannMT, StrongMA, HaoLY, GreiderCW (2001) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell107: 67–77
Google Scholar - KarlsederJ, BroccoliD, DaiY, HardyS, de LangeT (1999) p53‐ and ATM‐dependent apoptosis induced by telomeres lacking TRF2. Science283: 1321–1325
Google Scholar - LiuG, ParantJM, LangG, ChauP, Chavez‐ReyesA, El‐NaggarAK, MultaniA, ChangS, LozanoG (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet36: 63–68
Google Scholar - LoweSW, CeperoE, EvanG (2004) Intrinsic tumour suppression. Nature432: 307–315
Google Scholar - MaserRS, DePinhoRA (2002) Connecting chromosomes, crisis, and cancer. Science297: 565–569
Google Scholar - MeekerAK, HicksJL, Iacobuzio‐DonahueCA, MontgomeryEA, WestraWH, ChanTY, RonnettBM, De MarzoAM (2004) Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res10: 3317–3326
Google Scholar - MichaloglouC, VredeveldLC, SoengasMS, DenoyelleC, KuilmanT, van der HorstCM, MajoorDM, ShayJW, MooiWJ, PeeperDS (2005) BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature436: 720–724
Google Scholar - O’HaganRC, ChangS, MaserRS, MohanR, ArtandiSE, ChinL, DePinhoRA (2002) Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell2: 149–155
Google Scholar - RudolphKL, ChangS, LeeHW, BlascoM, GottliebGJ, GreiderC, DePinhoRA (1999) Longevity, stress response, and cancer in aging telomerase‐deficient mice. Cell96: 701–712
Google Scholar - TakaiH, SmogorzewskaA, de LangeT (2003) DNA damage foci at dysfunctional telomeres. Curr Biol13: 1549–1556
Google Scholar - ZhaoY, ChaiswingL, BakthavatchaluV, OberleyTD, St ClairDK (2006) Ras mutation promotes p53 activation and apoptosis of skin keratinocytes. Carcinogenesis27: 1692–1698
Google Scholar
Acknowledgements
We thank J. Karlseder for providing the TRF2_Δ_B_Δ_M retroviral vector. We acknowledge J. Ma and the T.C. Tsu Molecular Cytogenetics Core for outstanding cytogenetic services (NCI #CA016672). S.C. received generous financial support from the NCI (PO1 CA34936‐20), the Welch Medical Foundation, the Elsa U. Pardee Foundation, the Sidney Kimmel Foundation and the Michael Kadoorie Cancer Research Program.
Author information
Author notes
Wilfredo Cosme‐Blanco and Mei‐Feng Shen and Sen Pathak and Guillermina Lozano and Asha S Multani and Sandy Chang: Department of Cancer Genetics, The MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030, USA
Alexander J F Lazar: Departments of Pathology and Dermatology, The MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030, USA
- Alexander J F Lazar
Sandy Chang: Department of Hematopathology, The MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030, USA
- Sandy Chang
Authors
- Wilfredo Cosme‐Blanco
- Mei‐Feng Shen
- Alexander J F Lazar
- Sen Pathak
- Guillermina Lozano
- Asha S Multani
- Sandy Chang
Supplementary Information
Rights and permissions
Copyright: European Molecular Biology Organization
About this article
Cite this article
Cosme‐Blanco, W., Shen, M., Lazar, A.J.F. et al. Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53‐dependent cellular senescence.EMBO Rep 8, 497–503 (2007). https://doi.org/10.1038/sj.embor.7400937
- Received: 13 November 2006
- Revised: 05 February 2007
- Accepted: 06 February 2007
- Published: 30 March 2007
- Issue date: 01 May 2007
- DOI: https://doi.org/10.1038/sj.embor.7400937