Inferring sex-biased dispersal from population genetic tools: a review (original) (raw)
- Short Review
- Published: 25 March 2002
Heredity volume 88, pages 161–165 (2002) Cite this article
- 13k Accesses
- 381 Citations
- 8 Altmetric
- Metrics details
Abstract
Sex-biased dispersal, where individuals of one sex stay or return to their natal site (or group) to breed while individuals of the other sex are prone to disperse, is a wide-spread pattern in vertebrate organisms. In general, mammals exhibit male-biased dispersal whereas birds exhibit female-bias. Dispersal estimates are often difficult to obtain from direct field observations. Here we describe different methods for inferring sex-specific dispersal using population genetic tools and discuss the problems they can raise. We distinguish two types of methods: those based on bi-parental markers (eg comparison of male/female relatedness, F st and assignment probabilities) and those relying on the comparison between markers with different modes of inheritance (eg mtDNA markers and microsatellites). Finally, we discuss statistical problems that are encountered with these different methods (eg pseudoreplication, problems due to the comparison of distinct markers). While the genetic methods to detect sex-biased dispersal are now relatively well developed, their interpretation can prove problematic due to the confounding effects of factors such as the mating system of the species. Moreover, the relative power of these methods is not well known and requires further investigation.
Similar content being viewed by others
Introduction
Dispersal is a life history trait that has relevant effects on both the dynamics and the genetics of species. In this respect, the study of dispersal has been a major topic in evolutionary biology (Clobert et al, 2001). One specific pattern of dispersal, commonly called ‘sex-biased dispersal’, has received particular attention in field and theoretical studies during the past two decades (Pusey, 1987). Under this dispersal pattern, one sex is philopatric (ie individuals of this sex stay or return to their natal site or group to breed) while the other is more prone to disperse. For example, in mammals, dispersal is often male-biased (males have higher dispersal rates than females) (Dobson, 1982), whereas in birds the reverse pattern is generally found (Greenwood, 1980; Clarke et al, 1997).
Several hypotheses have been proposed to explain differential migration between sexes and can be broadly classified into three categories: (1) the ‘_resource-competition hypothesis_’ (Greenwood, 1980, 2) the ‘_local mate competition hypothesis_’ (Dobson, 1982; Perrin and Mazalov, 2000) and (3) the ‘_inbreeding avoidance hypothesis_’ (Pusey, 1987; Perrin and Mazalov, 2000). Predictions based on these different hypotheses depend on the mating system of the species considered. All three hypotheses predict male-biased dispersal in polygynous systems (the predominant mating system in mammals), whereas in monogamous systems (characteristic of birds), only the resource-competition model predicts a female-bias (Favre et al, 1997).
Understanding the evolutionary pressures leading to asymmetric dispersal of sexes and its ecological and genetic consequences will depend on our ability to determine its prevalence and magnitude in natural populations (Clarke et al, 1997; Aars and Ims, 2000). This is only possible if we have, at our disposal, effective, rapid and easy to use methods to detect sex-specific patterns of dispersal.
Slatkin (1985) distinguished two classes of methods for estimating levels of dispersal in natural populations. The first category, he called ‘direct methods’, comprises all approaches that rely on field observations measuring the extent of dispersal. While these methods can produce reliable results in certain circumstances, direct studies investigating dispersal (that use, for example, capture-release-recapture or radio-tracking methods) can be difficult to apply to certain types of organisms. Inferences of dispersal by direct methods may be misleading due to an inability to mark small organisms or to recapture a reasonable proportion of highly vagile organisms. In addition, death and emigration cannot be disentangled in such studies. Finally, direct estimates of migration can only measure mobility, that is the ability of an organism to move in space, and not the effective dispersal that reflects immigration followed by successful reproduction. Indeed, it is not always easy to verify subsequent reproduction of migrating individuals. This point is important because, in certain cases, effective dispersal can be low despite very high mobility (Gandon et al, 1997). The inverse is also possible, notably for sessile organisms whose gametes are mobile (Koenig et al, 1996).
Recent advances in genetic techniques (Slatkin's second class of methods), coupled with their increasing accessibility provide promising new opportunities to obtain ‘indirect estimates’ of dispersal. In addition, these methods can be applied without requiring intensive field observations (Neigel, 1997).
Recently, different authors have used population genetic tools for inferring sex-biased dispersal. However, the methodologies that have been proposed differ noticeably. In this paper, we describe these different methods and discuss the problems they can raise. We distinguish between two types of methods: methodologies based on bi-parentally inherited markers (eg allozymes, microsatellites) and those built from the comparisons between markers with different mode of inheritance (eg mtDNA, Y-linked markers, bi-parentally inherited markers). Finally, we discuss statistical problems that one can generally meets while undertaking such methods.
Before describing specifically the two types of methods, it seems important to note that there is a large difference between inferences based on bi-parentally inherited markers and those based on markers with different modes of inheritance. For the former, methods are generally based on the comparison between males and females after dispersal and before reproduction. In the offspring of these individuals, the sex-specific dispersal genetic signature does not exist any more since, for autosomal loci, all offspring inherit a randomly chosen allele from their father and mother. Therefore, if dispersal is no more sex-biased, in one generation the signal is lost. These methods are therefore only adequate to detect instantaneous (one generation) differences in dispersal. However, methods based on the comparison between different types of markers without reference to the sex will detect historical differences in gene-flow, even if it has disappeared today.
Inference from bi-parentally inherited markers
Different parameters were measured to infer sex differences in migration rates from bi-parentally inherited genetic markers. These methods generally rely on a particular sampling scheme of individuals in the sub-populations. As allele frequencies are equally randomized between males and females in the offspring, it is possible to detect contrasted population differentiation among sexes if individuals are sampled after dispersal. Therefore, for species in which dispersal occurs during the juvenile stage, it is optimal to sample adults and if possible not their progeny. Indeed, sampling the progeny will reduce the ability of the tests used to detect sex-biased dispersal.
Relatedness between individuals
Several authors have used the relatedness between individuals to infer dispersal differences between sexes (Ishibashi et al, 1997; Piertney et al, 1998; Knight et al, 1999; Surridge et al, 1999). Their logic can be summarised as follows: if individuals do not disperse far from the natal site (or group), one would expect individuals living in close proximity to be more related, on average, than individuals taken at random from the whole population. Therefore, in a situation where one sex is philopatric and the other sex is dispersing, individuals of the philopatric sex will have higher relatedness than those of the dispersing sex. In subdivided populations, it is possible to assess the relatedness between all pairs of individuals of each sex in each sub-population and test whether relatedness is higher for one sex than for the other. In instances where sub-populations are not well delimited, it is possible to test whether there is a relationship between relatedness and geographic distance for each sex and whether this relationship is the same for both (Knight et al, 1999). In all cases, the comparison between sexes potentially raises the statistical problem of pseudoreplication (ie the number of degrees of freedom is artificially increased) that we must take into account. We will return to this point below.
Fst
Another way to infer a bias in migration rates between males and females comes from the comparison of Wright's F st estimators computed both for males and females among sub-populations (Rassman et al, 1997; Balloux et al, 1998; Mossman and Waser, 1999). Indeed, F st is a parameter measuring the genetic differentiation (ie differences in allelic frequencies) between populations (Hartl and Clark, 1997). Thus, following the same logic as when using relatedness, one can expect that the sex with higher dispersal will have a lower between-sub-populations F st value compared with the sex that is dispersing less (eg Mossman and Waser, 1999).
Values of F st for males and females can be compared three ways. First, one can compute the global F st over all loci and sub-populations for each sex separately and generate confidence intervals by randomisation procedures (eg bootstrapping over loci, Goudet, 1995). If confidence intervals do not overlap for the two sexes, one can conclude that a significant difference exists (eg Mossman and Waser, 1999). The second way corresponds to the comparison of pairwise F st values (between each sub-population pair) obtained for each sex (Rassman et al, 1997). However, this latter method again potentially raises the problem of pseudoreplication (see below). Finally, Goudet et al (in press) have recently proposed a new method based on a randomisation procedure. The principle of this method is to generate independent random samples under the null hypothesis of no difference between males and females by randomly assigning a sex to individuals in each sub-population, but still respecting the observed sex ratio. A statistic (for example the difference between estimates of male and female F st) is calculated from each randomly created data set and is compared with the statistic obtained from the observed data set. The _P_-value of the test is then computed as the proportion of randomly obtained statistics with equal or more extreme values than the observed one (Goudet et al, in press). This method presents the advantage to eliminate pseudoreplication problems.
Vitalis (in press) has proposed another method to infer (and to quantify) sex-specific dispersal rates from F_st estimates. The author has shown, in particular, that the ratios of sex-specific F st parameters evaluated after dispersal over F st evaluated before dispersal are simple functions of sex-specific dispersal rates. Thus, the sex-specific migration rates can be estimated as 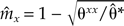 (for all X ∈ (♂♀}) where θ̂_xx is the multilocus estimator of F xx st among individuals of sex X sampled after dispersal and θ̂* is the multilocus estimator of F* st among individuals sampled before dispersal.
(for all X ∈ (♂♀}) where θ̂_xx is the multilocus estimator of F xx st among individuals of sex X sampled after dispersal and θ̂* is the multilocus estimator of F* st among individuals sampled before dispersal.
Assignment probabilities
The last method found in the literature was first applied by Favre et al (1997) to detect a bias in dispersal in the rodent species Crocidura russula. The authors used an ‘assignment index’ to determine the probability of a genotype originating from the population in which the individual was collected, as a way to distinguish potential immigrants from residents (Waser and Strobeck, 1998). An individual assignment index corresponds to the expected frequency of its genotype across all loci in the population in which it was collected (Paetkau et al, 1995; Waser and Strobeck, 1998). Because authors employing this method were not interested in population effects (which may arise from different levels of genetic diversity), assignment indices were corrected by subtracting assignment population means after log-transformation. Individuals with a strongly negative corrected assignment index have rare genotypes and thus are potentially immigrants or of recent immigrant ancestry. In terms of detecting sex-biased dispersal, Favre et al (1997) predicted that the more dispersing sex should have, on average, lower expected frequencies than the philopatric sex. Mossman and Waser (1999) used this method to detect a sex-biased dispersal in the whitefooted mouse, Peromyscus leucopus along with the more traditional method based on comparisons of F st.
From assignment indices, another method was developed to detect an asymmetric dispersal between sexes (Favre et al, 1997; Mossman and Waser, 1999). This method is based on the variance of corrected assignment indices. Variance is expected to be larger for the sex dispersing most (providing that not all individuals of one sex disperse).
With these methods, male and female parameters (mean or variance of corrected assignment indices) can also be compared using the randomization procedure proposed by Goudet et al (in press).
Power of the methods
The power of some of the genetic methods used to detect a sex-biased dispersal (F st, mean or variance of corrected assignment indices) has been recently tested by Goudet et al (in press) by computer simulations. From their results, many factors may influence the power of these tests, including dispersal rate, bias intensity and sampling scheme. It seems however that the two best methods (depending on population structure) are those based on F st (for high dispersal rates) and on the variance of corrected assignment indices (for low dispersal rates) (Goudet et al, in press).
Inference from the comparison between markers with different modes of inheritance
The comparison of genetic markers with different modes of inheritance, in particular sex-specific vs autosomal nuclear markers (eg allozymes, microsatellites), has constituted the principle method used to infer patterns of differential migration of sexes. This general approach has been applied to different vertebrate organisms including mammals (Melnick and Hoelzer, 1992; Baker et al, 1998; Seielstad et al, 1998; Pérez-Lezaun et al, 1999; Mesa et al, 2000; Escorza-Trevino and Dizon, 2000), birds (Guttierez, 1994; Gibbs et al, 2000; Piertney et al, 2000), reptiles (Karl et al, 1992; FitzSimmons et al, 1997; Rassmann et al, 1997) and fishes (Ferguson et al, 1993; Taylor et al, 1997; Patton et al, 1997). These different studies have employed several kinds of uni-parentally inherited markers (eg mtDNA, Y-linked markers). Although several Y- or W-linked genes have been identified in various mammalian and avian species respectively, few have proven to be sufficiently variable to be of particular utility in population genetics and thus were rarely employed for this type of problem (however, see studies on human dispersal and the use of short tandem repeats on the Y-chromosome, Seielstad et al, 1998; Pérez-Lezaun et al, 1999). In general, most molecular studies of sex-biased dispersal have relied on data from mtDNA in conjunction with those from nuclear markers.
The justification for the use of this comparative marker approach is that, for uni-parentally inherited markers, one sex does not contribute to a part of the genome of its offspring. In contrast, for bi-parental markers, both sexes contribute to the genetic diversity of the progeny. In this respect, differences in the level of genetic structure between the two kinds of markers are expected when sex-biased dispersal occurs. For species in which females are philopatric and males disperse, genetic differentiation between populations is expected to be higher when estimated using mtDNA (or another maternal marker) than using a bi-parental marker. This prediction is consistent with many genetic analyses of dispersal patterns in species known to have male-biased dispersal (eg Melnick and Hoelzer, 1992; FitzSimmons et al, 1997; Rassmann et al, 1997; Baker et al, 1998).
In the same way, differences in migration behaviour between sexes can also influence the level of genetic variation of paternally inherited DNA markers. Studies of human dispersal have shown a higher degree of genetic differentiation among populations using Y-chromosome genetic markers compared to mtDNA markers. These results have suggested a higher migration rate for females in human populations (Seielstad et al, 1998; Pérez-Lezaun et al, 1999).
Statistical analyses and problems raised
Although comparative marker approaches have been used in several studies, some authors have stressed the fact that results have to be interpreted carefully (eg Seielstad et al, 1998). Indeed, a difference in the level of genetic structure between different markers may be the outcome of their divergent mutation rates and/or effective population sizes (Chesser and Baker, 1996).
Mutation rate, in particular, can intervene in the degree of differentiation found between populations. Assuming an infinite island model, at equilibrium F st is equal to 1/[1 + 4_N_(m + μ)] in diploid systems (bi-parental markers) and 1/[1 + N(m + μ)] in haploid ones (uni-parentally inherited markers), where N is the effective population size, m the between-population migration rate and μ the mutation rate of the genetic marker. Mutation rates may be very different between different types of markers (eg Balloux et al, 2000) and may thus influence the level of observed genetic differentiation. To help avoid such problems, markers with similar mutation rates should ideally be used (eg Balloux et al, 2000).
The effective population size of haploid and diploid systems can also greatly influence the level of genetic structure observed for uni-parentally and bi-parentally inherited markers. It is generally assumed that the effective population size of haploid systems is four times less than in diploid systems in gonochoric species (see above) (Seielstad et al, 1998). However, Chesser and Baker (1996) have pointed out that this assumption is generally erroneous in natural populations due to a violation of the random mating assumption. Thus the effective population size of uni-parentally inherited genes will not always be smaller than diploid equivalents. The effective population size depends ultimately on the distribution of genetic diversity across sub-populations, which itself is a function of breeding characteristics (polygamy vs monogamy, size of the breeding group), of number of surviving progeny and of relative sex-specific dispersal levels. For example, under strong polygamy and low variance in reproductive success of females, the effective population size of bi-parentally inherited genes is smaller than that of maternally inherited genes (Chesser and Baker, 1996). Therefore, for the sake of accuracy, the estimate of male and female dispersal rates from estimates of differentiation provided by uni-parentally and bi-parentally inherited markers, requires the use of population genetic models that include the maximum information on the demographic parameters of the studied species (eg Petit et al, 2001).
For bi-parentally inherited markers, the comparison between male and female parameters (such as pairwise F st or between-individual relatedness, R) potentially raises a statistical problem of pseudoreplication. Indeed, the sample size n x (ie the total number of sub-populations or individuals analysed for each sex) is lower than the number of pairwise measures generated out of these data sets (there is n x(n x − 1)/2 parameter values for n x observations). This means that the number of degrees of freedom is artificially increased and therefore, in turn, the power of the test is similarly artificially increased.
This problem has rarely been taken into account. To our knowledge, only Knight et al (1999) have proposed statistical methods to avoid this caveat. Their analysis of dispersal patterns in Cichlid fishes was based on the use of a relatedness coefficient computed for each individual pair of each sex. They proposed two methods to account for pseudoreplication. For the first, a separate regression of relatedness coefficient (R) on geographic distance (D) was calculated for each individual. This resulted in n x individual regressions for each sex (where n x is the total number of individuals sampled for each sex and x ∈ {♂♀}). The slopes of all these individual regressions were then compared between sexes. The second approach was based on a comparison of means of ranked R-values. For each individual, the R-values (and corresponding D-values) were ranked in descending order and the means of ranked values were computed for both relatedness and distance. The slopes of the regression of the Rmean on Dmean obtained for males and females were then compared. This gave n x − 1 data points because an individual could not have an R-value with itself. Note that, for these two methods to be applied, the existence of isolation by distance is required. If this does not apply, other techniques must be employed.
Designing the comparison between sexes using data from any population no more than once or comparing the difference observed between sexes in our sample with the distribution generated by randomly assigning a sex to individuals in each sub-population respecting the observed sex ratio (Goudet et al, in press), are other possible way to eliminate pseudoreplication.
Conclusion
The degree of faithfulness to the natal site or social group is often sex-dependent. This specific dispersal pattern has received the name of sex-biased dispersal. In general, most mammalian species with asymmetric philopatry exhibit a male-biased dispersal, whereas most avian species exhibit a female biased dispersal. One likely consequence of such asymmetric dispersal rates is that a species will show different patterns of population structure for males and females. Sex-biased dispersal can therefore be detected by using the information contained in bi-parentally and/or uni-parentally inherited genetic markers.
While genetic methods to detect a bias in dispersal between sexes are now relatively well developed, their interpretation can prove problematic because of the influence of different factors such as the breeding structure of the studied species (notably for methods based on the comparison between markers with different modes of inheritance). Therefore, we recommend considerable caution in the interpretation of observed genetic patterns.
Finally, the genetic consequences of sex-biased dispersal at the within sub-population and metapopulation levels are still poorly documented. Theoretical predictions are now relatively well established, but we lack empirical data. Prout (1981) showed through an infinite island model that sex-biased dispersal results in a differentiation between males and females within each sub-population and thus leads to a heterozygous excess in the progeny. In this sense, sex-biased dispersal may act as a deterministic force counteracting the effects of inbreeding. Such predictions underline the importance of this life history trait in the ecological genetics of species. We hope that with the proper use of genetic inference methods, it will be possible to establish the general influence of sex-biased dispersal in the functioning of natural populations.
References
- Aars, J, Ims, RA (2000). Population dynamics and genetic consequences of spatial density-dependent dispersal in patchy populations. Am Nat, 155: 252–265.
PubMed Google Scholar - Baker, CS, Medrano-Gonzalez, L, Calambokidis, J, Perry, A, Pichler, F, Rosenbaum, H et al (1998). Population structure of nuclear and mitochondrial DNA variation among humpback whales in the North Pacific. Mol Ecol, 7: 695–707.
Article CAS PubMed Google Scholar - Balloux, F, Goudet, J, Perrin, N (1998). Breeding system and genetic variance in the monogamous, semi-social shrew, Crocidura russula. Evolution, 52: 1230–1235.
Article PubMed Google Scholar - Balloux, F, Brunner, H, Lugon-Moulin, N, Hausser, J, Goudet, J (2000). Microsatellites can be misleading: an empirical and simulation study. Evolution, 54: 1414–1422.
Article CAS PubMed Google Scholar - Chesser, RK, Baker, RJ (1996). Effective sizes and dynamics of uniparentally and diparentally inherited genes. Genetics, 144: 1225–1235.
CAS PubMed PubMed Central Google Scholar - Clarke, AL, Saether, B-E, Roskaft, E (1997). Sex biases in avian dispersal: a reappraisal. Oikos, 79: 429–438.
Article Google Scholar - Clobert, J, Nichols, JD, Danchin, E, Dhondt, A (2001). Dispersal. Oxford University Press: Oxford, UK.
Google Scholar - Dobson, FS (1982). Competition for mates and predominant juvenile male dispersal in mammals. Anim Behav, 30: 1183–1192.
Article Google Scholar - Escorza-Trevino, S, Dizon, AE (2000). Phylogeography, intraspecific structure and sex-biased dispersal of Dall's porpoise, Phocoenoides dalli, revealed by mitochondrial and microsatellite DNA analyses. Mol Ecol, 9: 1049–1060.
Article CAS PubMed Google Scholar - Favre, L, Balloux, F, Goudet, J, Perrin, N (1997). Female-biased dispersal in the monogamous mammal Crocidura russula: evidence from field and microsatellite patterns. Proc R Soc B, 269: 127–132.
Article Google Scholar - Ferguson, MM, Danzmann, RG, Arndt, SKA (1993). Mitochondrial-DNA and allozyme variation in Ontario cultured rainbow-trout spawning in different seasons. Aquaculture, 117: 237–259.
Article CAS Google Scholar - Fitzsimmons, NN, Moritz, C, Limpus, CJ, Pope, L, Prince, R (1997). Geographic structure of mitochondrial and nuclear gene polymorphisms in Australian green turtle populations and male-biased gene flow. Genetics, 147: 1843–1854.
CAS PubMed PubMed Central Google Scholar - Gandon, S, Ebert, D, Olivieri, I, Michalakis, Y (1997). Differential adaptation in spatially heterogenous environments and host-parasite coevolution. In: Mopper S, Strauss S (eds) Genetic Structure and Local Adaptation in Natural Insect Populations, Chapman & Hall: New York, pp 325–340.
Google Scholar - Gibbs, HL, Dawson, RJ, Hobson, KA (2000). Limited differentiation in microsatellite DNA variation among northern populations of the yellow warbler: evidence for male-biased gene flow? Mol Ecol, 9: 2137–2147.
Article CAS PubMed Google Scholar - Goudet, J (1995). FSTAT (vers. 1.2). A computer programm to calculate F-statistics. J Heredity, 86: 485–486.
Article Google Scholar - Goudet, J, Perrin, N, Waser, P (In press) Tests for sex biased dispersal using genetic markers. Mol Ecol.
- Greenwood, PJ (1980). Mating systems, philopatry and dispersal in birds and mammals. Anim Behav, 28: 1140–1162.
Article Google Scholar - Guttierez, PC (1994). Mitochondrial-DNA polymorphism in the oilbird (Steatornis-caripensis, steatornithidae) in Venezuela. Auk, 111: 573–578.
Google Scholar - Hartl, DL, Clark, AG (1997). Principles of Population Genetics, 3rd edn. Sinauer Associates: Sunderland, Massachusetts.
Google Scholar - Ishibashi, Y, Saitoh, T, Abe, S, Yoshida, MC (1997). Sex-related spatial kin structure in a spring population of grey-sided voles Clethrionomys rufocanus as revealed by mitochondrial and microsatellite DNA analyses. Mol Ecol, 6: 63–71.
Article CAS PubMed Google Scholar - Karl, SA, Bowen, BW, Avise, JC (1992). Global population genetic-structure and male-mediated gene flow in the green turtle (Chelonia mydas): RFLP analyses of anonymous nuclear loci. Genetics, 131: 163–173.
CAS PubMed PubMed Central Google Scholar - Knight, ME, Oppen, MJ, Smith, HL, Rico, C, Hewitt, GM, Turner, GF (1999). Evidence for male-biased dispersal in lake malawi cichlids from microsatellites. Mol Ecol, 8: 1521–1527.
Article CAS PubMed Google Scholar - Koenig, WD, Van Vuren, D, Hooge, PN (1996). Detectability, philopatry, and the distribution of dispersal distances in vertebrates. Trends Ecol Evol, 11: 514–517.
Article CAS PubMed Google Scholar - Melnick, DJ, Hoelzer, GA (1992). Differences in male and female macaque dispersal lead to contrasting distributions of nuclear and mitochondrial DNA variation. Int J Primatol, 13: 379–393.
Article Google Scholar - Mesa, NR, Mondragon, MC, Soto, ID, Parra, MV, Duque, C, Ortiz-Barrientos, D et al (2000). Autosomal, mtDNA, and Y-chromosome diversity in Amerinds: pre- and post-columbian patterns of gene flow in South America. Am J Hum Genet, 67: 1277–1286.
CAS PubMed PubMed Central Google Scholar - Mossman, CA, Waser, PM (1999). Genetic detection of sex-biased dispersal. Mol Ecol, 8: 1063–1067.
Article CAS PubMed Google Scholar - Neigel, JE (1997). A comparison of different alternative strategies for estimating gene flow from genetic markers. Ann Rev Ecol Syst, 28: 105–128.
Article Google Scholar - Paetkau, D, Calvert, W, Stirling, I, Strobeck, C (1995). Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol, 4: 347–354.
Article CAS PubMed Google Scholar - Patton, JC, Gallaway, BJ, Fechhelm, RG, Cronin, MA (1997). Genetic variation of microsatellite and mitochondrial DNA markers in broad whitefish (Coregonus nasus) in the Colville and Sagavanirktok Rivers in nothern Alaska. Can J Fish Aquat Sci, 54: 1548–1556.
Article CAS Google Scholar - Perez-Lezaun, A, Calafell, F, Comas, D, Mateu, E, Bosch, E, Martinez-Arias, R et al (1999). Sex-specific migration patterns in Central Asian Populations, revealed by analysis of Y-chromosome short tamdem repeats and mtDNA. Am J Hum Genet, 65: 208–219.
Article CAS PubMed PubMed Central Google Scholar - Perrin, N, Mazalov, V (2000). Local competition, inbreeding, and the evolution of sex-biased dispersal. Am Nat, 155: 116–127.
PubMed Google Scholar - Petit, E, Balloux, F, Goudet, J (2001). Sex-biased dispersal in a migratory bat: a characterization using sex-specific demographic parameters. Evolution, 55: 635–640.
Article CAS PubMed Google Scholar - Piertney, SB, Maccoll, AD, Bacon, PJ, Dallas, JF (1998). Local genetic structure in red grouse (Lagopus lagopus scoticus): evidence from microsatellite DNA markers. Mol Ecol, 7: 1645–1654.
Article CAS PubMed Google Scholar - Piertney, SB, Maccoll, AD, Bacon, PJ, Racey, PA, Lambin, X, Dallas, J (2000). Matrilineal genetic structure and female-mediated gene flow in red grouse (Lagopus lagopus scoticus): an analysis using mitochondrial DNA. Evolution, 54: 279–289.
CAS PubMed Google Scholar - Prout, T (1981). A note on the island model with sex dependent migration. Theor Appl Genet, 59: 327–332.
Article CAS PubMed Google Scholar - Pusey, A (1987). Sex-biased dispersal and inbreeding avoidance in birds and mammals. Trends Ecol Evol, 2: 295–299.
Article CAS PubMed Google Scholar - Rassmann, K, Tautz, D, Trillmich, F, Gliddon, C (1997). The microevolution of the Galapagos marine iguana Amblyrchynchus cristalus assessed by nuclear and mitochondrial genetic analyses. Mol Ecol, 6: 437–452.
Article CAS Google Scholar - Seielstad, MT, Minch, E, Cavalli-Sforza, LL (1998). Genetic evidence for a higher female migration rate in humans. Nat Genet, 20: 278–280.
Article CAS PubMed Google Scholar - Slatkin, M (1985). Gene Flow in natural populations. Ann Rev Ecol Syst, 16: 393–430.
Article Google Scholar - Surridge, AK, Ibrahim, KM, Bell, DJ, Webb, NJ, Rico, C, Hewitt, GM (1999). Fine-scale genetic structuring in a natural population of European wild rabbits (Oryctolagus cuniculus). Mol Eco, 8: 299–307.
Article CAS Google Scholar - Taylor, EB, Harvey, S, Pollard, S, Volpe, J (1997). Postglacial genetic differentiation of reproductive ecotypes of kokanee Oncorhynchus nerka in Okanagan Lake, British Columbia. Mol Ecol, 6: 503–517.
Article CAS PubMed Google Scholar - Vitalis, R (In press) Sex-specific genetic differentiation and coalescence times: estimating sex-biased dispersal rates. Mol Ecol
- Waser, PM, Strobeck, C (1998). Genetic signature of interpopulation dispersal. Trends Ecol Evol, 13: 43–44.
Article CAS PubMed Google Scholar
Acknowledgements
We thank F Balloux, J Goudet and K MacKoy for very useful discussions and suggestions. We also thank two anonymous reviewers. This work received financial support from the CNRS (PNDBE) and the MENRT (PRFMMIP no 95).
Author information
Authors and Affiliations
- Centre d'Etude du Polymorphisme des Micro-organismes, UMR 9926 CNRS-IRD, Institut de Recherche et Développement, 911 av. Agropolis BP 5045, Montpellier, Cedex 1, 34032, France
F Prugnolle & T de Meeus
Authors
- F Prugnolle
- T de Meeus
Corresponding author
Correspondence toF Prugnolle.
Rights and permissions
About this article
Cite this article
Prugnolle, F., de Meeus, T. Inferring sex-biased dispersal from population genetic tools: a review.Heredity 88, 161–165 (2002). https://doi.org/10.1038/sj.hdy.6800060
- Received: 16 August 2001
- Accepted: 09 October 2001
- Published: 25 March 2002
- Issue date: 01 March 2002
- DOI: https://doi.org/10.1038/sj.hdy.6800060