Prenatal Infection and Risk for Schizophrenia: IL-1β, IL-6, and TNFα Inhibit Cortical Neuron Dendrite Development (original) (raw)
- Original Article
- Published: 07 April 2004
Preclinical Research
Neuropsychopharmacology volume 29, pages 1221–1229 (2004)Cite this article
- 4660 Accesses
- 211 Citations
- Metrics details
Abstract
Prenatal exposure to infection increases risk for schizophrenia, and we have hypothesized that inflammatory cytokines, generated in response to maternal infection, alter neuron development and increase risk for schizophrenia. We sought to study the effect of cytokines generated in response to infection—interleukin-1_β (IL-1_β), tumor necrosis factor-α (TNF_α_), and interleukin-6 (IL-6)—on the dendritic development of cortical neurons. Primary mixed neuronal cultures were obtained from E18 rats and exposed to 0, 100, or 1000 units (U)/ml of IL-1_β_, TNF_α_, IL-6, or IL-1_β_+TNF_α_ for 44 h. MAP-2-positive neurons were randomly identified for each condition and the number of primary dendrites, nodes, and total dendrite length was determined. We found that 100 U of TNF_α_ significantly reduced the number of nodes (27%, p_=0.02) and total dendritic length (14%, p_=0.04), but did not affect overall neuron survival. A measure of 100 U IL-1_β+TNF_α significantly reduced the number of primary dendrites (17%, _p_=0.006), nodes (32%, _p_=0.001), and total dendritic length (30%, p<0.0001), although it did not affect overall neuron survival. At 1000 U, each cytokine significantly reduced the number of primary dendrites (14–24%), nodes (28–37%), as well as total dendritic length (25–30%); neuron survival was reduced by 14–21%. These results indicate that inflammatory cytokines can significantly reduce dendrite development and complexity of developing cortical neurons, consistent with the neuropathology of schizophrenia. These findings also support the hypothesis that cytokines play a key mechanistic role in the link between prenatal exposure to infection and risk for schizophrenia.
Similar content being viewed by others
INTRODUCTION
Maternal infection during pregnancy increases the risk of the offspring developing schizophrenia and other neurodevelopmental disorders. The weight of the evidence indicates that maternal influenza infection during pregnancy is associated with a higher incidence of schizophrenia in offspring (reviewed in McGrath and Murray, 2003; Bagalkote et al, 2001). Other types of maternal infections have also been implicated, including pneumonia and diphtheria (Watson et al, 1984; O’Callaghan et al, 1994), rubella (Brown et al, 2000b), measles, varicella-zoster, and polio (Torrey et al, 1988; Suvisaari et al, 1999). Recent studies have moved beyond epidemiologic association with studies that link infections in individual mothers with schizophrenia in their adult children. Respiratory infections in the second trimester increase risk for schizophrenia (Brown et al, 2000a). In this same cohort, serologic evidence of maternal exposure to influenza also increased the risk of schizophrenia in offspring (Brown et al, 2004a). Finally, the offspring of mothers with elevated IgG and IgM levels, and antibodies to herpes simplex virus type 2 during pregnancy, have an increased risk for schizophrenia (Buka et al, 2001a).
The pathological mechanisms responsible for increased risk of schizophrenia in offspring after maternal infection remain largely unstudied. Hypotheses about the role of infection in the etiology of schizophrenia have focused on direct infection of the developing fetus (Yolken and Torrey, 1995) or the generation of antibodies that crossreact with neuronal antigens (Wright et al, 1993). As a variety of infections are associated with increased risk of schizophrenia, a feature common to all infections would be a likely candidate mechanism. We have hypothesized that cytokines generated in response to maternal infection alter early brain development and increase risk for schizophrenia (Gilmore and Jarskog, 1997). Cytokines, including interleukin-1_β_ (IL-1_β_), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF_α_), have a variety of effects on the central nervous system (CNS) and are expressed by glial and neuronal elements within the CNS (Bartfai and Schultzberg, 1993; Hopkins and Rothwell, 1995; Rothwell and Hopkins, 1995). Cytokines, including IL-1_β_, IL-6, and TNF_α_, regulate normal brain development and have been implicated in abnormal brain development (Merrill, 1992; Mehler and Kessler, 1994; Mehler and Kessler, 1997). Expression of cytokine mRNA in the CNS is developmentally regulated in mouse, rat, sheep, and human brain (Burns et al, 1993; Gadient and Otten, 1994; Pousset, 1994; Mousa et al, 1999; Dziegielewska et al, 2000), an indication of the important role that cytokines play in neurodevelopment.
IL-1_β_, IL-6, and TNF_α_ are elevated in the maternal–fetal unit after maternal infection in human pregnancies (Hillier et al, 1993; Fortunato et al, 1996; Yoon et al, 2003) and in animal models (Fidel et al, 1994; Urakubo et al, 2001). Maternally generated cytokines cross the placenta and regulate cell growth and development in the fetus (Medlock et al, 1993; Letterio et al, 1994; Stallmach and Karolyi, 1994; Li et al, 1995; McDuffie et al, 2001). An additional source of cytokines in prenatal infection may be the placenta, as the human placenta synthesizes IL-1_β_, IL-6, and TNF_α_ in response to infection (Fortunato et al, 1996; Taniguchi et al, 1991; Menon et al, 1995). Finally, the fetus itself can mount an inflammatory response, especially of IL-6, in the face of maternal infection (Gomez et al, 1998; Yoon et al, 2003). IL-1_β_, IL-6, and TNF_α_ cross the blood–brain barrier in mature rodents (Banks et al, 1991; Guiterrez et al, 1993; Banks et al, 1994). Finally, the blood–brain barrier is incomplete in the fetus (Adinolfi, 1985), making it very likely that systemically generated cytokines gain entry into the fetal brain.
Several recent studies support the hypothesis that cytokines play a key role in the association between maternal infection, altered brain development, and risk for schizophrenia. Maternal blood levels of TNF_α_ (Buka et al, 2001b) and IL-8 (Brown et al, 2004b) are elevated in pregnancies in which the offspring goes on to develop schizophrenia. Three animal models of maternal infection have recently been advanced as models of schizophrenia that also support our hypothesis. Maternal infection with human influenza virus in mice causes abnormalities in prepulse inhibition (Shi et al, 2003), and maternal exposure to Escherichia coli cell wall endotoxin, lipopolysaccharide (LPS), disrupts sensorimotor gating in the offspring (Borrell et al, 2002). Finally, maternal exposure to poly-I:C, a synthetic double-stranded RNA that stimulates a cytokine response, causes prepulse inhibition abnormalities (Shi et al, 2003) and disrupted latent inhibition (Zuckerman and Weiner, 2003; Zuckerman et al, 2003). In the influenza model, no virus is detected in the fetal brain (Shi et al, 2003), suggesting that the immune responses, especially cytokines, are the likely mediators of the abnormal brain development that leads to long-term behavioral changes. In the poly-I:C and LPS models, it is likely that the immune response to the challenge plays a major role in the mechanism of action, as no infectious agent is present. We have shown that maternal LPS exposure increases cytokine expression in the placenta and amniotic fluid of rats (Urakubo et al, 2001).
Cytokines can be neurotoxic to developing neurons, as they decrease survival of serotonergic and dopaminergic neurons (Jarskog et al, 1997), hippocampal neurons (Araujo and Cotman, 1995), and cortical neurons (Gelbard et al, 1993; Jeohn et al, 1998; Marx et al, 2001). The neuropathology of cerebral cortex in schizophrenia is subtle and does not appear to involve neuron loss, but rather loss of neuropil (Selemon et al, 2003), dendrites and spines (Glantz and Lewis, 2000; Broadbelt et al, 2002), and synaptic markers (Glantz and Lewis, 1997). The actions of cytokines on developmental processes that may give rise to schizophrenia, such as dendrite development, are less well known. This study was conducted to determine the effect of cytokines on cortical neuron dendrite development. We hypothesized that the inflammatory cytokines IL-1_β_, IL-6, and TNF_α_, which are generated in response to maternal infection, would decrease the development of dendrites on embryonic cortical neurons in vitro.
MATERIALS AND METHODS
Cell Culture
Primary cortical neuronal cells were isolated as described previously (Marx et al, 2001). Briefly, frontal cerebral cortex was dissected from whole brains of embryonic day 18 Sprague–Dawley rats (Charles River, Raleigh, NC) and the meninges removed. The trimmed tissues were dissociated in 0.125% trypsin and 0.5 mM EDTA for 20 min at 37°C. The reaction was terminated by adding an equal volume of Dulbecco's modified Eagle's medium+10% fetal bovine serum+5 units (U)/ml penicillin/streptomycin. The cells were further dissociated by mechanical trituration, followed by filtering through the nylon mesh and centrifugation. The cells (5 × 104/ml/well) were plated on 18-mm glass coverslips coated with poly-D-lysine (100 μg/ml) in 12-well culture plates. Cells were cultured in Neurobasal medium supplemented with B27 (Invitrogen, Carlsbad, CA), 0.5 mM L-glutamine, 25 μM L-glutamate and 5 U/ml penicillin/streptomycin. After 4 h, media was exchanged and cultures were treated with TNF_α_, IL-6 (R&D Systems, Minneapolis, MN), IL-1_β_ (Boehringer Mannheim, Ridgefield, CT), or TNF_α_+IL-1_β_ for 44 h at concentrations of 0, 100, or 1000 U/ml, three wells per condition. Specific activity per manufacturer: TNF_α_, 60 U/ng; IL-6, 12.5 U/ng; and IL-1_β_, 50 U/ng. Three cultures from three different litters were studied for a total _n_=9 per treatment condition.
Immunohistochemistry
After 44 h of cytokine exposure, cultures were rinsed in Hank's balanced salt solution, fixed in 4% paraformaldehyde, and permeabilized with 0.2%. Triton X-100 in phosphate-buffered saline (PBS) and incubated overnight with goat polyclonal antibody MAP-2 (1 : 100). To confirm that neurons in culture expressed the respective cytokine receptors, three cover glasses each were incubated overnight with goat polyclonal antibodies for TNF receptor-1 (1 : 100) and TNF receptor-2 (1 : 100), or rabbit polyclonal IL-1 receptor-1 (1 : 500) or IL-6 receptor-α (1 : 200). MAP-2 and cytokine receptor antibodies were purchased from Santa Cruz (Santa Cruz, CA). To determine if glial cells were present, cultures were incubated overnight with monoclonal mouse anti-GFAP (Boehringer Mannheim, 1 : 500). Cultures were then processed using the avidin–biotin peroxidase complex (ABC) method (Vector). Following immunochemistry and osmium tetraoxide intensification, cultures were rinsed in PBS and counterstained with toluidine blue. A negative control experiment was also performed to rule out nonspecific staining in the absence of primary antibodies. In this experiment, ABC immunochemistry procedures were followed, but no primary antibody was included.
Cell Counting and Dendrite Morphology
Primary cortical neurons were cultured at low density (5 × 104 cells/ml/well) to allow morphological analysis of single neurons. Neuronal survival was analyzed by counting neurite-bearing MAP-2-immunostained cells with clear neuronal morphology at × 200 magnification using an ocular grid. In all, 12 grid areas were examined per coverslip using a blinded, randomized procedure (0.25 mm2 per grid area × 12 per coverslip=total sample area of 3.00 mm2 per coverslip). Cell counts were converted to percentages of intraexperimental controls.
For morphometry, the first 10 single neurons that had two or more dendrites encountered from the center of the coverslip were digitally captured at × 60. Dendrites of scanned cells were traced using Neurolucida and NeuroExplorer software (Micro Bright Field, Williston, VT). Morphometry data from individual neurons were collected for a number of primary dendrites per neuron, branch points per neuron, and total dendrite length per neuron (Figure 1). Primary dendrites were defined as neurites originating at the neuronal somata. A total of 90 neurons were studied for each treatment condition.
Figure 1
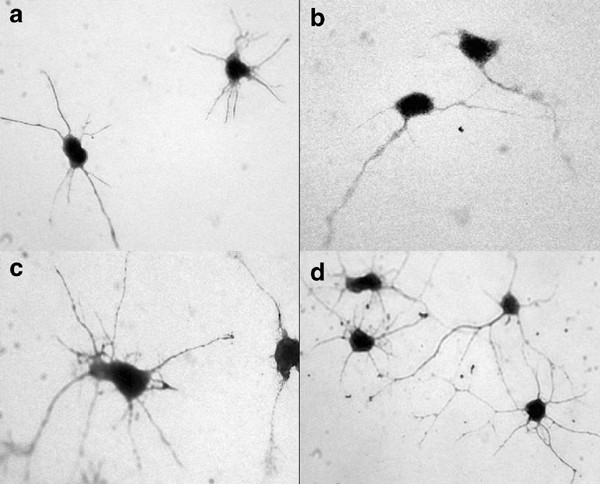
Immunodetection of receptors for IL-1_β_, IL-6, and TNF_α_ on embryonic cortical neurons in vitro using antibodies to (a) IL-1r, (b) IL-6r, (c) TNFr1, and (d) TNFr2 ( × 60 magnification).
Data Analysis
_T_-tests and one-way analysis of variance (ANOVA) with post hoc Dunnett's multiple comparison tests were performed. Significance was set at p<0.05 using a two-tailed test for all analyses.
RESULTS
Cortical neurons in culture expressed receptors for IL-1, IL-6, and TNF (types 1 and 2); Figure 1). There were no GFAP-positive cells present in the cultures. Representative MAP-2-positive neurons and their dendrite tracings are presented in Figure 2. At a concentration of 100 U, TNF_α_ significantly reduced the number of nodes (27%, p_=0.02) and total dendrite length (14%, p_=0.04; Table 1) of MAP-2-labeled cortical neurons in culture. IL-1_β+TNF_α (100 U) significantly reduced the number of primary dendrites (17%, _p_=0.006), nodes (32%, _p_=0.001), and total dendrite length (30%, p<0.0001; Table 1). There were no significant effects of IL-1_β_ or IL-6 on the number of primary dendrites, nodes, or total dendrite length at this concentration. At 100 U, there were no significant effects of IL-1_β_, IL-6, TNF_α_, or IL-1_β_+TNF_α_ on neuron survival (_p_>0.05 for each cytokine).
Figure 2
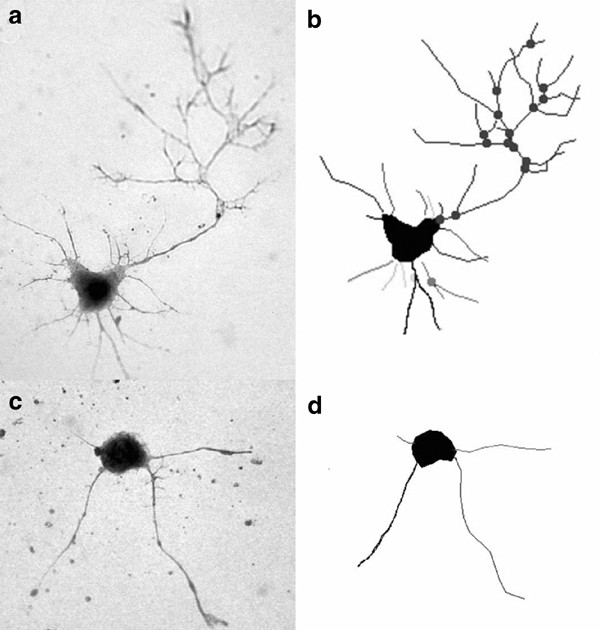
Representative MAP-2-labeled neurons and their dendrite tracings from neurolucida: (a and b) control; (c and d) 100 U of IL-1_β_+TNF_α_ ( × 60 magnification).
Table 1 Morphometric analysis of cortical neurons in primary culture treated with 100 U of IL-1_β_, IL-6, TNF_α_, or IL-1_β_+TNF_α_ for 44 h
At a concentration of 1000 U, IL-1_β_ significantly reduced the number of primary dendrites (14%, p<0.05), nodes (37%, p<0.01), as well as total dendrite length (30%, p<0.01; Figure 3) on cortical neurons. TNF_α_ also significantly reduced the number of primary dendrites (24%, p<0.01), nodes (29%, p<0.01), and total dendrite length (27%, p<0.01). IL-6 significantly reduced the number of primary dendrites (16%, p<0.05), nodes (33%, p<0.01), and total dendrite length (25%, p<0.01). Finally, IL-1_β_+TNF_α_ significantly reduced the number of primary dendrites (17%, p<0.01), nodes (28%, p<0.05), and total dendrite length (29%, p<0.01). At 1000 U, there was a small but significant decrease in neuronal survival for IL-1_β_ (14%, p<0.05), IL-6 (14%, p<0.05), TNF_α_ (17%; p<0.05), and IL-1_β_+TNF_α_ (22%, p<0.01; Figure 4).
Figure 3
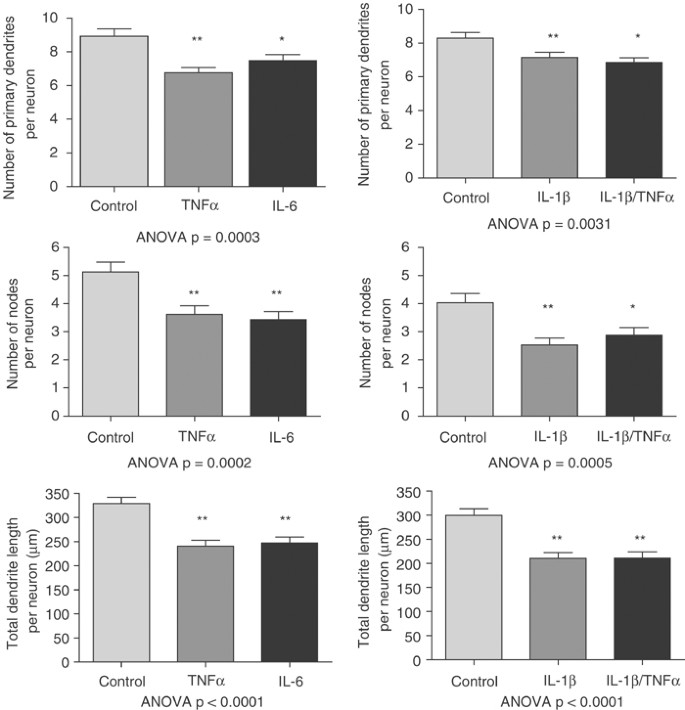
Morphometric analysis of cortical neurons in primary culture treated with 1000 U of IL-1_β_, IL-6, TNF_α_, or IL-1_β_+TNF_α_ for 44 h. Each cytokine significantly decreased primary dendrite number, number of nodes, and total dendrite length. Results of ANOVA are presented with each experiment. Post hoc Dunnett's test: *p<0.05; **p<0.01.
Figure 4
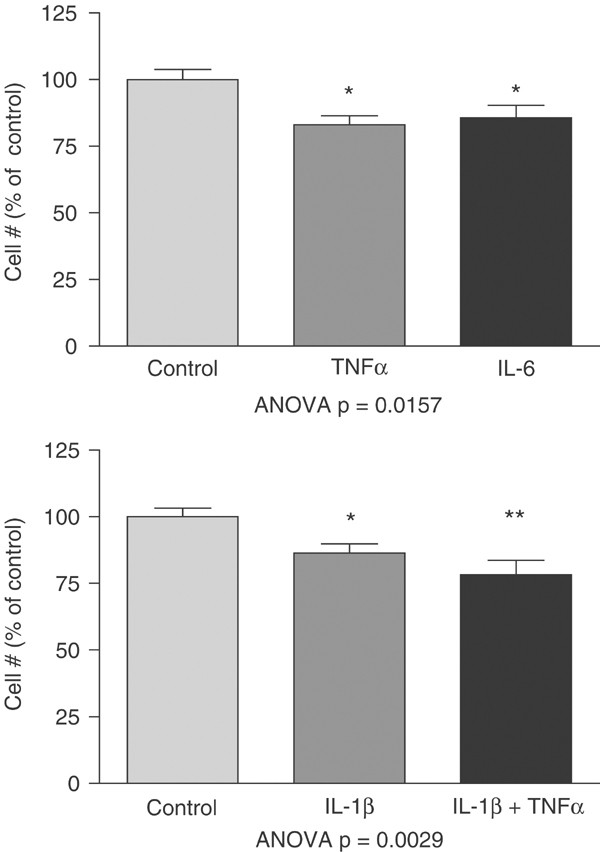
Neuron survival after exposure to 1000 U of IL-1_β_, IL-6, TNF_α_, or IL-1_β_+TNF_α_ for 44 h. Each cytokine significantly decreased overall neuron survival. Results of ANOVA presented with each experiment. Post hoc Dunnett's test: *p<0.05; **p<0.01. A measure of 100 U of each cytokine had no significant effect on neuron survival.
DISCUSSION
This study demonstrates that the inflammatory cytokines IL-1_β_, IL-6, and TNF_α_, which are typically generated in response to infection, can significantly inhibit the development of dendrites in embryonic cortical neurons. TNF_α_ (100 U) decreases the number of nodes and total dendrite length; the combination of IL-1_β_ and TNF_α_ at 100 U causes a robust decrease in the number of primary dendrites, nodes, and total dendrite length, suggesting a synergistic effect. At the higher dose (1000 U), IL-1_β_, IL-6, and TNF_α_ caused a significant reduction in each parameter of dendrite morphology. These findings are consistent with a recent study reporting that TNF_α_ significantly decreased neurite outgrowth and branching of hippocampal neurons in culture (Neumann et al, 2002). Interferon-γ also inhibits dendritic growth in hippocampal neurons (Kim et al, 2002). The synergistic effect of IL-1_β_ and TNF_α_ on dendrite morphology is also consistent with a previous study that found that IL-1_β_ and TNF_α_ had an additive effect on decreased cortical neuron survival (Jeohn et al, 1998).
While it is difficult to compare directly studies due to variations in assay methods and in the unit activity of recombinant cytokines, the concentrations of cytokines used in this experiment are within physiologic ranges. A measure of 100 U is approximately 2000 pg/ml of TNF_α_, 8000 pg /ml of IL-6, and 1666 pg/ml of IL-1_β_. In a previous study, we found levels of TNF_α_, IL-6, IL-1_β_ in the fetal rat brain to be 1500 pg/g, 1500 pg/g, and 5000 pg/g tissue, respectively (Urakubo et al, 2001). Brain IL-1 β levels in adult rats and mice after peripheral LPS exposure reach levels of 50 000 pg/g (Nguyen et al, 1998) and 5000 pg/g of tissue for IL-6 (Meyer et al, 1997). Levels in human CSF in the setting of infection range as high as 8000 pg/ml for IL-1_β_ in CSF after aseptic meningitis (Ramilo et al, 1990) and 26 000 pg/ml of TNF_α_ in CSF after bacterial meningitis (Mustafa et al, 1989). Local concentrations of cytokines at a neuronal level are likely to be much higher.
The decreased dendrite development of cortical neurons observed after cytokine exposure offers a tangible link between an important environmental risk factor for schizophrenia and the cortical neuropathology observed in schizophrenia. Prenatal exposure to maternal infection, through the generation of systemic cytokines, could decrease dendritic development of cortical neurons, which would ultimately lead to the ‘miswiring’ and ‘dysconnectivity’ of cortical circuits thought to underlie the clinical and cognitive symptoms of schizophrenia. Especially relevant is the lowest concentration tested, in which 100 U TNF_α_ and the combination of IL-1_β_ and TNF_α_ caused significant decreases in dendrite development while having no effect on overall neuron survival. This finding is consistent with the cortical neuropathology of schizophrenia that involves decreased neuropil (Selemon et al, 2003), dendrites and dendritic spines (Glantz and Lewis, 2000; Broadbelt et al, 2002), and synaptic markers (Glantz and Lewis, 1997), but no neuronal loss (Selemon et al, 2003).
As noted above, studies in human pregnancies (Hillier et al, 1993; Fortunato et al, 1996; Yoon et al, 2003) and in animal models (Fidel et al, 1994; Urakubo et al, 2001) indicate that maternal infection increases systemic levels of IL-1_β_, IL-6, and TNF_α_. The source of systemic cytokines can be the maternal immune system, the placenta, and even the fetus itself. Prenatal exposure to maternal infection can also activate microglia and astrocytes in the developing cortex (Cai et al, 2000; Fatemi et al, 2002). These activated glial cells would be a potential local source of cytokines and may play a role in the transduction of the immune response to maternal infection to developing neurons. We have recently shown that maternal infection can also regulate the expression of brain-derived neurotrophic factor and nerve growth factor in the fetal and neonatal brain (Gilmore et al, 2003). Neurotrophic factors are expressed by glial cells and represent an additional mechanism through which maternal infection can regulate neuronal development.
Apoptosis represents another potential molecular mechanism by which altered expression of cytokines and neurotrophic factors could alter cortical neuronal development. Proinflammatory cytokines are well-recognized activators of neuronal apoptosis (Hu et al, 1997) and recent data implicate apoptosis in several disorders of neurodevelopment including schizophrenia (Jarskog et al, 2004). While activation of apoptosis is often associated with cell death, emerging evidence indicates that apoptotic activity can also underlie neuronal atrophy (Ona et al, 1999), neuritic degeneration (Ivins et al, 1998), and synaptic loss (Gilman and Mattson, 2002) without causing cell death. Certain apoptotic regulatory proteins are developmentally expressed in patterns that appear to increase the susceptibility to pathological apoptosis during early neurodevelopment (Hu et al, 2000; Jarskog and Gilmore, 2000; Yakovlev et al, 2001). Taken together, these data suggest that an apoptotic mechanism can contribute to cytokine-mediated effects on altered cortical development in schizophrenia and other neurodevelopmental disorders.
_β_-catenin is a member of the Wnt signaling pathway and has recently been shown to also regulate dendrite morphology, an action that involves interactions between β_-catenin, cadherin, the actin cytoskeleton, and the cell membrane (Yu and Malenka, 2003). IL-6 and TNF_α can decrease _β_-catenin expression in hepatocarcinoma cells (Cervello et al, 2001) and in bronchial epithelial cells (Carayol et al, 2002), suggesting that cytokines may act through a mechanism that involves β_-catenin in developing dendrites. As noted above, TNF_α significantly decreased dendrite development of hippocampal neurons; this effect involved a Rho GTPase-dependent mechanism that also regulates the cytoskeleton (Neumann et al, 2002). These studies suggest that regulation of the cytoskeleton is a potential mechanism through which cytokines can inhibit the development of dendrites.
While little is known about the impact of infection on the developing cortex, there has been much interest in the role that cytokines play in the white matter damage associated with cerebral palsy. Intrauterine infection greatly increases the risk of periventricular leukomalacia (PVL) and cerebral palsy in premature infants and term infants (Grether and Nelson, 1997; Wu and Colford, 2000; Wu, 2002), and cytokines have been implicated in this association (Dammann and Leviton, 1997). Elevated levels of IL-6 in fetal cord blood and of IL-6 and IL-1_β_ in amniotic fluid are associated with white matter lesions and cerebral palsy in premature infants (Yoon et al, 1996; Yoon et al, 2000). Children with cerebral palsy have high neonatal blood levels of cytokines, including IL-1_β_ and TNF_α_ (Nelson et al, 1998). Finally, premature infants with cerebral lesions on MRI exhibit high levels of IL-1_β_, IL-6, IL-10, and TNF_α_ in the cord blood at birth (Duggan et al, 2001). In addition to elevations of cytokines in fetal blood and amniotic fluid, there is evidence that IL-1_β_, IL-6, and TNF_α_ are highly expressed in white matter around PVL lesions, especially in astrocytes and microglial cells (Deguchi et al, 1996; Yoon et al, 1996; Kadhim et al, 2001). A recent study indicates that there is increased expression of IL-1_β_ and TNF_α_ by cortical and subcortical neurons in the brains of infants with PVL (Kadhim et al, 2001), indicating that the same inflammatory processes that lead to PVL may also alter cortical neuron development.
These studies with regard to the connection between cytokines and PVL offer clues as to how prenatal exposure to infection might affect the developing brain and increase risk for schizophrenia. Gross white matter damage is the only one of many possible outcomes of prenatal exposure to infection that likely includes more subtle alterations of cortical neuron and/or white matter development. Gross white matter lesions are not more frequently seen in schizophrenia (Rivkin et al, 2000), although more subtle abnormalities of white matter volume and diffusion tensor properties are present (Sigmundsson et al, 2001; Kubicki et al, 2002). Very little is known about the impact of prenatal exposure to infection on brain development beyond white matter, although maternal influenza infection can cause abnormal corticogenesis in mice (Fatemi et al, 1999). Our study provides additional evidence that maternal infection can alter cortical neuronal development as well.
The ultimate impact of infection on the developing brain is probably dependent on the severity and timing of the infection in relation to specific neurodevelopmental events, as well as the genetic susceptibility of the individual. For example, genes that regulate the immune response, especially in the setting of prenatal exposure to infection, would likely have an effect on early brain development. Studies have found associations between schizophrenia and polymorphisms of cytokine genes that increase cytokine production, including TNF_α_ (Boin et al, 2001; Jun et al, 2003; Meira-Lima et al, 2003) and IL-1_β_ (Katila et al, 1999). Further, there are associations between polymorphisms for IL-1_β_ (Meisenzahl et al, 2001) and the TNF receptor-II (Wassink et al, 2000) and abnormal brain morphology in patients with schizophrenia, including lateral ventricle enlargement and cortical gray matter and white matter reductions.
Infection during pregnancy can be associated with other mechanisms that could also adversely affect prenatal brain development, including stress hormones (Trejo et al, 1995; Koenig et al, 2002), hypoxia (Altshuler, 1993), reduced blood flow to the fetus (Altshuler, 1993), malnutrition (Butler et al, 1994), and hyperthermia (Upfold and Smith, 1988). Ultimately, a full mechanistic understanding of the impact of maternal infection on the fetal brain development will have to integrate knowledge about the involvement of inflammatory, hormonal, and other potential mechanisms.
Finally, inflammatory cytokines play a mechanistic role in other forms of insults to the developing brain, including hypoxia–ischemia (Szaflarski et al, 1995; Grow and Parks, 2002) and excitotoxic injury (Silverstein et al, 1997). It has also been shown that infection sensitizes the immature brain to hypoxic–ischemic injury, suggesting a combined activation of inflammatory mediators in the brain (Eklind et al, 2001). Cytokines therefore may represent a final common pathway for the variety of perinatal complications that are associated with increased risk for schizophrenia (Cannon et al, 2002). The study of perinatal complications and schizophrenia is often criticized for being too nonspecific with regard to the mechanisms that underlie a diverse group of complications. Inflammatory cytokines offer an attractive unifying mechanism.
In summary, the inflammatory cytokines IL-1_β_, IL-6, and TNF_α_ each decrease dendritic development of cortical neurons in vitro, consistent with the cortical neuropathology observed in schizophrenia. Schizophrenia is likely the result of a complex interaction between multiple genetic and environmental risk factors. If one assumes that schizophrenia is a disorder of synaptic connectivity, any molecule or process that regulates dendritic growth and synapse formation in the developing brain may contribute to risk. Given the evidence that prenatal exposure to maternal infection, hypoxia, and other insults activate an immune response in the developing brain, inflammatory cytokines are emerging as an important candidate mechanism in understanding pre- and perinatal risk factors for schizophrenia. The study of interactions between the immune system and the developing brain is just beginning, but offers a key to understanding one of the etiologic factors contributing to schizophrenia and other neurodevelopmental disorders. This line of investigation may ultimately offer targets for therapeutic strategies that may prevent or reduce the risk of abnormal brain development in the setting of infection and other perinatal insults, and may ultimately prevent or reduce the risk of schizophrenia.
References
- Adinolfi A (1985). The development of the human blood–CSF–brain barrier. Dev Med Child Neurol 27: 532–537.
CAS PubMed Google Scholar - Altshuler G (1993). Some placental considerations related to neurodevelopmental and other disorders. J Child Neurol 8: 78–94.
CAS PubMed Google Scholar - Araujo DM, Cotman CW (1995). Differential effects of interleukin-1b and interleukin-2 on glia and hippocampal neurons in culture. Int J Dev Neurosci 13: 201–212.
CAS PubMed Google Scholar - Bagalkote H, Pang D, Jones PB (2001). Maternal influenza and schizophrenia in offspring. Int J Ment Health 39: 3–21.
Google Scholar - Banks WA, Kastin AJ, Guiterrez EG (1994). Penetration of interleukin-6 across the murine blood–brain barrier. Neurosci Lett 179: 53–56.
CAS PubMed Google Scholar - Banks WA, Ortiz L, Plotkin SR, Kastin AJ (1991). Human interleukin (IL) 1_α_, murine IL-1_α_ and murine IL-1_β_ are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther 259: 988–996.
CAS PubMed Google Scholar - Bartfai T, Schultzberg M (1993). Cytokines in neuronal cell types. Neurochem Int 22: 435–444.
CAS PubMed Google Scholar - Boin F, Zanardini R, Pioli R, Altamura CA, Maes M, Gennarelli M (2001). Association between −G308A tumor necrosis factor alpha gene polymorphism and schizophrenia. Mol Psychiatry 6: 79–82.
CAS PubMed Google Scholar - Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C (2002). Prenatal immune challenge disrupts sensorimotor gating in adult rats: implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology 26: 204–215.
CAS PubMed Google Scholar - Broadbelt K, Byne W, Jones LB (2002). Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res 58: 75–81.
PubMed Google Scholar - Brown AS, Begg M, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M et al (2004a). Serologic evidence for prenatal influenza in the etiology of schizophrenia. Am J Psychiatry, (in press).
- Brown AS, Cohen P, Greenwald S, Susser E (2000b). Nonaffective psychosis after exposure to rubella. Am J Psychiatry 157: 438–443.
CAS PubMed Google Scholar - Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V et al (2004b). Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Arch Gen Psychiatry, (in press).
- Brown AS, Schaefer CA, Wyatt RJ, Goetz R, Begg M, Gorman JM et al (2000a). Maternal exposure to respiratory infections and adult schizophrenia spectrum disorders: a prospective birth cohort study. Schizophr Bull 26: 287–295.
CAS PubMed Google Scholar - Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Bernstein D, Yolken RH (2001a). Maternal infections and subsequent psychosis among offspring. Arch Gen Psychiatry 58: 1032–1037.
CAS PubMed Google Scholar - Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH (2001b). Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun 15: 411–420.
CAS PubMed Google Scholar - Burns TM, Clough JA, Klein RM, Wood GW, Berman NEJ (1993). Developmental regulation of cytokine expression in the mouse brain. Growth Factors 9: 253–258.
CAS PubMed Google Scholar - Butler PD, Susser ES, Brown AS, Kaufman CA, Gorman JM (1994). Prenatal nutritional deprivation as a risk factor in schizophrenia: preclinical evidence. Neuropsychopharmacology 11: 227–235.
CAS PubMed Google Scholar - Cai Z, Pan Z-L, Pang Y, Evans OB, Rhodes PG (2000). Cytokine induction in fetal rat brain and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res 47: 64–72.
CAS PubMed Google Scholar - Cannon M, Jones PB, Murray RM (2002). Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 159: 1080–1092.
PubMed Google Scholar - Carayol N, Campbell A, Vachier I, Mainprice B, Bousquet J, Godard P et al (2002). Modulation of cadherin and catenins expression by tumor necrosis factor-α and dexamethasone in human bronchial epithelial cells. Am J Respir Mol Cell Biol 26: 341–347.
CAS Google Scholar - Cervello M, Notarbartolo M, Landion M, Cusimano A, Virruso L, Montalto G et al (2001). Downregulation of wild-type _β_-catenin expression by interleukin 6 in hepatocarcinoma HepG2 cells: a possible role in the growth-regulatory effects of a cytokine. Eur J Cancer 37: 512–519.
CAS PubMed Google Scholar - Dammann O, Leviton A (1997). Maternal intrauterine infection, cytokines, and brain damage in the premature infant. Pediatr Res 42: 1–8.
CAS PubMed Google Scholar - Deguchi K, Mizuguchi M, Takashima S (1996). Immunohistochemical expression of tumor necrosis factor-α in neonatal leukomalacia. Pediatr Neurol 14: 13–16.
CAS PubMed Google Scholar - Duggan PJ, Maalouf EF, Watts TL, Sullivan MHF, Counsell SJ, Allsop J et al (2001). Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet 358: 1699–1700.
CAS PubMed Google Scholar - Dziegielewska KM, Moller JE, Potter AM, EK J, Lane MA, Sanders NR (2000). Acute-phase cytokines IL-1_β_ and TNF-α in brain development. Cell Tissue Res 299: 335–345.
CAS PubMed Google Scholar - Eklind S, Mallard C, Leverin AL, Gilland E, Mattsby-Baltzer I, Hagberg H (2001). Bacterial endotoxin sensitizes the immature brain to hypoxic–ischemic injury. Eur J Neurosci 13: 1101–1106.
CAS PubMed Google Scholar - Fatemi SH, Emamian ES, Kist DA, Sidwell RW, Nakajima K, Akhter P et al (1999). Defective corticogenesis and reduction in reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry 4: 145–154.
CAS PubMed Google Scholar - Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA et al (2002). Human influenza virus infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry 7: 633–640.
CAS PubMed Google Scholar - Fidel Jr PL, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H et al (1994). Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. Am J Obstet Gynecol 170: 1467–1475.
CAS PubMed Google Scholar - Fortunato SJ, Menon RP, Swan KF, Menon R (1996). Inflammatory cytokines (interleukins 1,6,8 and tumor necrosis factor-α) release from cultured fetal membranes in response to endotoxic lipopolysaccharide mirrors amniotic fluid. Am J Obstet Gynecol 174: 1855–1862.
CAS PubMed Google Scholar - Gadient RA, Otten U (1994). Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Res 637: 10–14.
CAS PubMed Google Scholar - Gelbard HA, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG (1993). Neurotoxic effects of tumor necrosis factor in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci 15: 417–422.
CAS PubMed Google Scholar - Gilman CP, Mattson MP (2002). Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromol Med 2: 197–214.
CAS Google Scholar - Gilmore JH, Jarskog LF (1997). Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res 24: 365–367.
CAS PubMed Google Scholar - Gilmore JH, Jarskog LF, Vadlamudi S (2003). Maternal infection alters BDNF and NGF expression in the fetal and neonatal brain and maternal–fetal unit of the rat. J Neuroimmunol 138: 49–55.
CAS PubMed Google Scholar - Glantz LA, Lewis DA (1997). Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia: regional and diagnostic specificity. Arch Gen Psychiatry 54: 943–952.
CAS PubMed Google Scholar - Glantz LA, Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65–73.
CAS PubMed Google Scholar - Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM (1998). The fetal inflammatory syndrome. Am J Obstet Gynecol 179: 194–202.
CAS PubMed Google Scholar - Grether JK, Nelson KB (1997). Maternal infection and cerebral palsy in infants of normal birth weight. JAMA 278: 207–211.
CAS PubMed Google Scholar - Grow J, Parks JD (2002). Pathogenesis of hypoxic–ischemic cerebral injury in the term infant: current concepts. Clin Perinatol 29: 585–602.
PubMed Google Scholar - Guiterrez EG, Banks WA, Kastin AJ (1993). Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J Neuroimmunol 47: 169–176.
Google Scholar - Hillier SL, Witkin SS, Krohn MA, Watts DH, Kiviat NB, Eschenbach DA (1993). The relationship of amniotic fluid cytokines and preterm delivery, amniotic fluid infection, histologic chorioamnionitis, and chorioamnion infection. Am J Obstet Gynecol 81: 941–948.
CAS Google Scholar - Hopkins SJ, Rothwell NJ (1995). Cytokines and the nervous system I: expression and recognition. Trends Neurosci 18: 83–88.
CAS PubMed Google Scholar - Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjo BK (2000). Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab 20: 1294–1300.
CAS PubMed Google Scholar - Hu S, Peterson PK, Chao CC (1997). Cytokine-mediated neuronal apoptosis. Neurochem Int 30: 427–431.
CAS PubMed Google Scholar - Ivins KJ, Bui ETN, Cotman CW (1998). Amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis 5: 365–378.
CAS PubMed Google Scholar - Jarskog LF, Gilmore JH (2000). Developmental expression of Bcl-2 protein in human cortex. Dev Brain Res 119: 225–230.
CAS Google Scholar - Jarskog LF, Selinger ES, Lieberman JA, Gilmore JH (2004). Apoptotic proteins in temporal cortex in schizophrenia: high Bax/Bcl-2 ratio without caspase-3 activation. Am J Psychiatry 161: 109–115.
PubMed Google Scholar - Jarskog LF, Xiao H, Wilkie MB, Lauder JM, Gilmore JH (1997). Cytokine regulation of embryonic dopaminergic and serotonergic neuron survival in vitro. Int J Dev Neurosci 15: 711–716.
CAS PubMed Google Scholar - Jeohn GH, Kong LY, Wilson B, Hong J (1998). Synergistic neurotoxic effects of combined treatments with cytokines in murine primary mixed neuron/glia cultures. J Neuroimmunol 85: 1–10.
CAS PubMed Google Scholar - Jun T-Y, Pae C-U, Chae J-H, Bahk W-M, Kim K-S, Han H et al (2003). TNF_β_ polymorphism may be associated with schizophrenia in the Korean population. Schizophr Res 61: 39–45.
PubMed Google Scholar - Kadhim H, Tabarki B, De Prez C, Sebrire G (2001). Cytokine immunoreactivity in cortical and subcortical neurons in periventricular leukomalacia: are cytokines implicated in neuronal dysfunction in cerebral palsy? Acta Neuropathol 105: 209–216.
Google Scholar - Katila H, Hanninen K, Hurme M (1999). Polymorphisms of the interleukin-1 gene complex in schizophrenia. Mol Psychiatry 4: 179–181.
CAS PubMed Google Scholar - Kim I-J, Beck HN, Lein PJ, Higgins D (2002). Interferon γ induces retrograde dendritic retraction and inhibits synapse formation. J Neurosci 22: 4530–4539.
CAS PubMed PubMed Central Google Scholar - Koenig JI, Kirkpatrick B, Lee P (2002). Glucocorticoid hormones and early brain development in schizophrenia. Neuropsychopharmacology 27: 309–318.
CAS PubMed Google Scholar - Kubicki M, Westin C-F, Maier SE, Frumin M, Nestor PG, Salisbury DF et al (2002). Uncinate fasciculus findings in schizophrenia: a magnetic resonance diffusion tensor imaging study. Am J Psychiatry 159: 813–820.
PubMed PubMed Central Google Scholar - Letterio JJ, Geiser AG, Kulkarni AB, Roche NS, Sporn MB, Roberts AB (1994). Maternal rescue of transforming growth factor-_β_1 null mice. Science 264: 36–38.
Google Scholar - Li Y, Ohls RK, Rosa C, Shah M, Richards DS, Christensen RD (1995). Maternal and umbilical serum concentrations of granulocyte colony-stimulating factor and its messenger RNA during clinical chorioamnionitis. Obstet Gynecol 86: 428–432.
CAS PubMed Google Scholar - Marx CE, Jarskog LF, Lauder JM, Lieberman JA, Gilmore JH (2001). Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for schizophrenia. Biol Psychiatry 50: 743–749.
CAS PubMed Google Scholar - McDuffie RS, Dabies JK, Leslie KK, Sherman MP, Gibbs RS (2001). A randomized control trial of interleuklin-1 receptor antagonist in a rabbit model of ascending infection in pregnancy. Infect Dis Obstet Gynecol 9: 233–237.
CAS PubMed PubMed Central Google Scholar - McGrath J, Murray R (2003). Risk factors for schizophrenia: from conception to birth. In Hirsh SR, Weinberger DR (eds). Schizophrenia. Blackwell Science: Malden, MA. pp 232–250.
Google Scholar - Medlock ES, Kaplan DL, Cecchini M, Ulich TR, del Castillo J, Andresen J (1993). Granulocyte-stimulating factor crosses the placenta and stimulates fetal granulopoiesis. Blood 81: 916–922.
CAS PubMed Google Scholar - Mehler MF, Kessler JA (1994). Growth factor regulation of neuronal development. Dev Neurosci 16: 180–195.
CAS PubMed Google Scholar - Mehler MF, Kessler JA (1997). Hematolymphopoietic and inflammatory cytokines in neural development. Trends Neurosci 20: 357–365.
CAS PubMed Google Scholar - Meira-Lima IV, Pereira AC, Mota GF, Floriano M, Araujo F, Mansur AJ et al (2003). Analysis of a polymorphism of the promoter region of the tumor necrosis factor alpha gene in schizophrenia and bipolar disorder: further support for an association with schizophrenia. Mol Psychiatry 8: 718–720.
CAS PubMed Google Scholar - Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G et al (2001). Association of an IL-1_β_ genetic polymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry 158: 1316–1319.
CAS PubMed Google Scholar - Menon R, Swan KF, Lyden TW, Rote NS, Fortunato SJ (1995). Expression of inflammatory cytokines (interleukin-1_β_ and interleukin-6) in amniochorionic membranes. Am J Obstet Gynecol 172: 493–500.
CAS PubMed Google Scholar - Merrill JE (1992). Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: normal and pathological. Dev Neurosci 14: 1–10.
CAS PubMed Google Scholar - Meyer TA, Wang JJ, Tiao GM, Ogle CK, Fischer JE, Hasselgren PO (1997). Sepsis and endotoxemia in mice stimulate the expression of interleukin-1 and interleukin-6 in the central nervous system. Clin Sci 92: 519–525.
CAS Google Scholar - Mousa A, Seiger A, Kjaeldgaard A, Bakhiet M (1999). Human first trimester forebrain cells express genes for inflammatory and anti-inflammatory cytokines. Cytokine 11: 55–60.
CAS PubMed Google Scholar - Mustafa MM, Lebel MH, Ramilo O, Olsen KD, Reisch JS, Beutler B et al (1989). Correlation of interleukin-1 beta and cachectin concentrations in cerebrospinal fluid and outcome from bacterial meningitis. J Pediatr 115: 208–213.
CAS PubMed Google Scholar - Nelson KB, Dambrosia JM, Grether JK, Phillips TM (1998). Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol 44: 665–675.
CAS PubMed Google Scholar - Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde Y-A (2002). Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a Rho-dependent mechanism. J Neurosci 22: 854–862.
CAS PubMed PubMed Central Google Scholar - Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR et al (1998). Exposure to acute stress induces brain interleukin-1_β_ protein in the rat. J Neurosci 18: 2239–2246.
CAS PubMed PubMed Central Google Scholar - O’Callaghan E, Sham PC, Talei N, Murray G, Glover G, Murray RM (1994). The relationship of schizophrenic births to 16 infectious diseases. Br J Psychiatry 165: 353–356.
PubMed Google Scholar - Ona VO, Li M, Vonsattel JPG, Andrews LJ, Khan SQ, Chung WM et al (1999). Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399: 263–267.
CAS PubMed Google Scholar - Pousset F (1994). Developmental expression of cytokine genes in the cortex and hippocampus of the rat central nervous system. Dev Brain Res 81: 143–146.
CAS Google Scholar - Ramilo O, Mustafa MM, Porter J, Saez-Llorens X, Mertsola J, Olsen KD et al (1990). Detection of interleukin 1 beta but not tumor necrosis factor-alpha in cerebrospinal fluid of children with aseptic meningitis. Am J Dis Child 144: 349–352.
CAS PubMed Google Scholar - Rivkin P, Kraut M, Barta P, Anthony J, Arria AM, Pearlson G (2000). White matter hyperintensity volume in late-onset and early-onset schizophrenia. Int J Geriatr Psychiatry 15: 1085–1089.
CAS PubMed Google Scholar - Rothwell NJ, Hopkins SJ (1995). Cytokines and the nervous system II: actions and mechanisms of actions. Trends Neurosci 18: 130–136.
CAS PubMed Google Scholar - Selemon LD, Mrzljak J, Kleinman JE, Herman MM, Goldman-Rakic PS (2003). Regional specificity in the neuropathologic substrates of schizophrenia. Arch Gen Psychiatry 60: 69–77.
PubMed Google Scholar - Shi L, Fatemi SH, Sidewell RW, Patterson PH (2003). Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 23: 297–302.
PubMed PubMed Central Google Scholar - Sigmundsson T, Suckling J, Maier M, Williams SCR, Bullmore ET, Greenwood KE et al (2001). Structural abnormalities in frontal, temporal, and limbic regions and interconnecting white matter tracts in schizophrenia in schizophrenic patients with prominent negative symptoms. Am J Psychiatry 158: 234–243.
CAS PubMed Google Scholar - Silverstein FS, Barks JD, Hagan P, Liu XH, Ivacko J, Szaflarski J (1997). Cytokines and perinatal brain injury. Neurochem Int 30: 375–383.
CAS PubMed Google Scholar - Stallmach T, Karolyi L (1994). Augmentation of fetal granulopoiesis with chorioamnionitis during the second trimester of gestation. Hum Pathol 25: 244–247.
CAS PubMed Google Scholar - Suvisaari J, Haukka J, Tanskanen A, Hovi T, Lonnqvist J (1999). Association between prenatal exposure to poliovirus infection and adult schizophrenia. Am J Psychiatry 156: 1100–1102.
CAS PubMed Google Scholar - Szaflarski J, Burtrum D, Silverstein FS (1995). Cerebral hypoxia–ischemia stimulates cytokine gene expression in perinatal rats. Stroke 26: 1093–1100.
CAS PubMed Google Scholar - Taniguchi T, Matsuzaki N, Kameda T, Shimoya K, Jo T, Saji F et al (1991). The enhanced production of placental interleukin-1 during labor and intrauterine infection. Am J Obstet Gynecol 165: 131–137.
CAS PubMed Google Scholar - Torrey EF, Rawlings R, Waldman IN (1988). Schizophrenic births and viral diseases in two states. Schizophr Res 1: 73–77.
CAS PubMed Google Scholar - Trejo JL, Machin C, Arahuetes RM, Rua C (1995). Influence of maternal adrenalectomy and glucocorticoid administration on the development of rat cerebral cortex. Anat Embryol 192: 89–99.
CAS Google Scholar - Upfold JB, Smith MSR (1988). Maternal hyperthermia as a cause of ‘idiopathic’ mental retardation. Med Hypotheses 27: 89–92.
CAS PubMed Google Scholar - Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH (2001). Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res 47: 27–36.
CAS PubMed Google Scholar - Wassink TH, Crowe RR, Andreasen NC (2000). Tumor necrosis factor receptor-II: heritability and effect on brain morphology in schizophrenia. Mol Psychiatry 5: 678–682.
CAS PubMed Google Scholar - Watson CG, Kucala T, Tilleskjor C, Jacobs L (1984). Schizophrenic birth seasonality in relation to the incidence of infectious diseases and temperature extremes. Arch Gen Psychiatry 41: 85–90.
CAS PubMed Google Scholar - Wright P, Gill M, Murray RM (1993). Schizophrenia: genetics and the maternal immune response to viral infection. Am J Med Genet (Neuropsychiatr Genet) 48: 40–46.
CAS Google Scholar - Wu YW (2002). Systemic review of chorioamnionitis and cerebral palsy. Ment Retard Dev Disabilities Res Rev 8: 25–29.
Google Scholar - Wu YW, Colford JM (2000). Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. JAMA 284: 1417–1424.
CAS PubMed Google Scholar - Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K et al (2001). Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci 21: 7439–7446.
CAS PubMed PubMed Central Google Scholar - Yolken RH, Torrey EF (1995). Viruses, schizophrenia, and bipolar disorder. Clin Microbiol Rev 8: 131–145.
CAS PubMed PubMed Central Google Scholar - Yoon BH, Romero R, Moon J, Chaiworapongsa T, Espinoza J, Kim YM et al (2003). Differences in the fetal interleukin-6 response to microbial invasion of the amniotic cavity between term and preterm gestation. J Maternal–Fetal Neonatal Med 13: 32–38.
CAS Google Scholar - Yoon BH, Romero R, Park JS, Kim JC, Kim YM, Choi J-H et al (2000). Fetal exposure to an intra-amniotic inflammation and the development of cerebral palsy at the age of three years. Am J Obstet Gynecol 182: 675–681.
CAS PubMed Google Scholar - Yoon BH, Romero R, Yang SH, Jun JK, Kim IO, Choi JH et al (1996). Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am J Obstet Gynecol 174: 1433–1440.
CAS PubMed Google Scholar - Yu X, Malenka RC (2003). Catenin is critical for dendritic morphogenesis. Nat Neurosci 6: 1169–1177.
CAS PubMed Google Scholar - Zuckerman L, Rehavi M, Weiner I (2003). Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 25: 1778–1789.
Google Scholar - Zuckerman L, Weiner I (2003). Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacology 169: 308–313.
CAS PubMed Google Scholar
Acknowledgements
This work was supported by MH 60352 (JHG) and MH 01752 (LFJ).
Author information
Authors and Affiliations
- UNC Schizophrenia Research Center, Chapel Hill, NC, USA
John H Gilmore & Lars Fredrik Jarskog - Department of Psychiatry, Chapel Hill, NC, USA
John H Gilmore, Lars Fredrik Jarskog & Swarooparani Vadlamudi - Department of Cell and Developmental Biology University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Jean M Lauder
Authors
- John H Gilmore
You can also search for this author inPubMed Google Scholar - Lars Fredrik Jarskog
You can also search for this author inPubMed Google Scholar - Swarooparani Vadlamudi
You can also search for this author inPubMed Google Scholar - Jean M Lauder
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJohn H Gilmore.
Rights and permissions
About this article
Cite this article
Gilmore, J., Fredrik Jarskog, L., Vadlamudi, S. et al. Prenatal Infection and Risk for Schizophrenia: IL-1_β_, IL-6, and TNF_α_ Inhibit Cortical Neuron Dendrite Development.Neuropsychopharmacol 29, 1221–1229 (2004). https://doi.org/10.1038/sj.npp.1300446
- Received: 18 February 2004
- Revised: 18 February 2004
- Accepted: 22 February 2004
- Published: 07 April 2004
- Issue Date: 01 July 2004
- DOI: https://doi.org/10.1038/sj.npp.1300446