Dopamine is not Required for the Hyperlocomotor Response to NMDA Receptor Antagonists (original) (raw)
INTRODUCTION
Antagonists of the _N_-methyl-D-aspartate (NMDA) receptor can produce psychotic symptoms in humans that are indistinguishable from acute episodes of schizophrenia (Snyder, 1976; Javitt and Zukin, 1991; Steinpreis and Salamone, 1993) and exacerbate existing psychoses in schizophrenics (Javitt and Zukin, 1991). As a result, glutamatergic dysfunction has been proposed as an underlying cause of the pathophysiology of schizophrenia (Kim et al, 1980; Javitt and Zukin, 1991; Olney and Farber, 1995; Coyle, 1996). Hence, NMDA receptor antagonists such as phencyclidine (PCP), dizocilpine maleate (MK-801), and ketamine have been used extensively to pharmacologically model psychosis in animals. Many of the behaviors produced in rodents treated with NMDA receptor antagonists have relevance to symptoms of schizophrenia in humans. These include disruptions in prepulse inhibition and deficits in working memory (Bakshi et al, 1994; Verma and Moghaddam, 1996). Subanesthetic doses of drugs such as PCP and MK-801 also cause stereotyped motor behaviors and increased forward locomotion (Sturgeon et al, 1979; Moghaddam and Adams, 1998; Andine et al, 1999). Although positive symptoms of schizophrenia cannot be directly studied in animals, rodents exhibit substantial locomotor activation in response to NMDA receptor antagonists. As the neural circuitry mediating positive symptoms in humans and locomotor activation in rodents may be partly overlapping, we have focused on the locomotor-stimulating effects of NMDA receptor antagonists.
Motor behaviors are often associated with an activated dopamine system (Creese and Iversen, 1973; Kelly et al, 1975), and increased dopamine transmission is thought to be a major factor in the psychotomimetic actions of PCP, MK-801, and related drugs. Systemic administration of NMDA receptor antagonists results in an increase in both dopamine and glutamate efflux in limbic brain regions such as the nucleus accumbens (NAc) and prefrontal cortex, presumably through a disinhibitory mechanism (Imperato et al, 1990; Miller and Abercrombie, 1996; Adams and Moghaddam, 1998). Systemically administered PCP increases the firing rate of dopaminergic neurons (Freeman and Bunney, 1984; French et al, 1993), and dopamine receptor antagonists, including antipsychotic drugs such as haloperidol and clozapine, attenuate PCP-induced stereotypies and locomotion (Sturgeon et al, 1981; Freed et al, 1984). Finally, PCP-induced hyperlocomotion can be reversed by 6-hydroxydopamine (6-OHDA) lesions of the NAc (French and Vantini, 1984; French et al, 1985; Steinpreis and Salamone, 1993). Together these findings suggest that dopamine is necessary for the behavioral effects of NMDA receptor antagonists, and are consistent with a hyperdopaminergic hypothesis of schizophrenia (for review, see Davis et al, 1991).
However, there is also evidence that dopamine is not required for the actions of NMDA receptor antagonists, but that increases in dopamine levels may be an indirect consequence of activated glutamatergic transmission at non-NMDA receptors (Takahata and Moghaddam, 2003). In support of this, the behavioral effects of PCP correlate more closely with glutamate release in the NAc and prefrontal cortex than with dopamine release (Adams and Moghaddam, 1998). A systemically administered group II metabotropic glutamate receptor (mGlu 2/3) agonist, LY354740, prevents PCP-induced glutamate release in the prefrontal cortex and NAc without affecting dopamine release, and it reduces PCP-induced locomotor activity, stereotypy, and working memory deficits (Moghaddam and Adams, 1998). Some studies have shown that behavioral effects of PCP and MK-801, including locomotor stimulation and potentiation of cocaine reinforcement, can occur without concomitant increases in dopamine release (Druhan et al, 1996; Pierce et al, 1997; Cornish et al, 2001). Rats can be trained to self-administer both PCP and MK-801 in the presence of a dopamine antagonist, suggesting that the rewarding effects of these drugs are dopamine-independent (Carlezon and Wise, 1996). NMDA antagonists have also been shown to produce hyperlocomotion in mice and rats after temporary catecholamine depletion by reserpine (Carlsson and Carlsson, 1989a) or alpha-methyl-para-tyrosine (Lapin and Rogawski, 1995; Swanson and Schoepp, 2002). Taken together, the existing evidence leaves open the question of whether dopamine is required for the psychotomimetic behavioral effects of NMDA receptor antagonists. Most studies that demonstrate effects independent of dopamine are performed in animals with intact dopamine systems, making it difficult to conclude that dopamine is not involved. While lesioning experiments are compelling, they are hampered by the fact that complete lesions result in hypophagia and subsequent death (Ungerstedt, 1971). Incomplete lesions, coupled with resultant hypersensitivity to dopamine (Ungerstedt et al, 1974), have made it difficult to definitively rule out a contribution from dopamine.
Here, we use a mouse model in which mice are genetically depleted of dopamine (dopamine-deficient (DD) mice) to test the role of dopamine in the hyperlocomotive response to NMDA receptor antagonists. In DD mice, the gene encoding tyrosine hydroxylase is inactivated specifically in dopaminergic neurons (Zhou and Palmiter, 1995). DD mice are hypoactive and aphagic, and die 3–4 weeks postnatally if not rescued by daily injections of the synthetic dopamine precursor L-3,4-dihydroxyphenylalanine (L-dopa), which temporarily restores dopamine synthesis and regulated release within dopaminergic neurons (Zhou and Palmiter, 1995; Szczypka et al, 1999). Dopamine levels in the striatum of DD mice reach approximately 9% of wild-type levels 3 h after L-dopa treatment, but fall to <1% within 24 h (Szczypka et al, 1999). DD mice are supersensitive to dopamine, as treatment with the daily regimen of L-dopa causes hyperlocomotion and induces striatal c-fos expression—effects not seen in control mice (Kim et al, 2000). Using this model, along with pharmacological blockade of glutamate release, we were able to test whether dopaminergic and glutamatergic signaling are required for the locomotor-stimulating effects of PCP and MK-801. In addition, we were able to test the effects of PCP and MK-801 in DD mice after introduction of small amounts of releasable dopamine by pretreatment with a low dose of L-dopa. This allowed us to determine whether dopamine can modulate the locomotor response to NMDA antagonists, something that cannot be easily performed in other models of dopamine depletion.
MATERIALS AND METHODS
Generation and Maintenance of DD Mice
DD mice were created as described by inactivating both tyrosine hydroxylase (Th) alleles and subsequently targeting a Th gene to the dopamine β hydroxylase (Dbh) locus (Zhou and Palmiter, 1995). The Th −/− ; Dbh Th/+ pups representing DD mice were identified at 2–3 weeks of age by their runted appearance and behavioral responsiveness to L-dopa. The mice were maintained in a modified specific-pathogen-free facility. DD mice were kept alive by once daily injections of L-dopa (50 mg/kg/day). This dose of L-dopa elicits locomotor activity in DD mice that is similar in magnitude each day (Zhou and Palmiter, 1995; Kim et al, 2000). Control mice were littermates of DD mice that had at least one intact Th gene and one intact Dbh gene; these mice produce nearly normal levels of dopamine and norepinephrine (Thomas et al, 1998; Rios et al, 1999). L-Dopa treatment of control mice has no effect on locomotor activity, striatal dopamine levels, or Fos immunoreactivity (Szczypka et al, 1999; Kim et al, 2000), and was therefore not administered to control mice in these studies. Purina chow (5LJ5) and water were available ad libitum except during experimental procedures. All procedures were conducted in accordance with guidelines established by the National Institutes of Health and the University of Washington Animal Care Committee.
Drugs
PCP (phencyclidine hydrochloride) (Sigma, St Louis, MO), MK-801 (dizocilpine hydrogen maleate) (Sigma), SCH 23390 hydrochloride (Sigma), haloperidol (Sigma), and LY 379268 (gift from Dr Darryl Schoepp, Eli Lilly Company) were dissolved in phosphate-buffered saline (PBS; 10 mM phosphate, 150 mM NaCl, pH 7.0). L-Dopa (1.5 mg/ml; Sigma) was dissolved in PBS containing 2.5 mg/ml ascorbic acid. Vehicle control was PBS alone. All treatments were by intraperitoneal (i.p.) injection in a volume of 10 μl/g body weight, except L-dopa, which is injected at 33 μl/g body weight.
Behavioral Analysis
Locomotor activity was measured in transparent Plexiglas cages (40 × 20 × 20 cm) set in activity chambers equipped with four infrared beams set 8.8 cm apart (San Diego Instruments Inc., San Diego, CA). The number of consecutive beam breaks is reported as ‘ambulations’. All animals were allowed to acclimate to the chambers for at least 12 h prior to beginning a test so that novelty of the testing apparatus was not a factor in the behavioral response to drug administration. Locomotor tests with DD mice were performed 18 h after their last L-dopa (50 mg/kg) injection. Each drug-treatment day was followed by at least a 3-day washout period, during which DD mice were treated only with L-dopa (50 mg/kg) and control mice were left untreated. Because of the limitations of breeding and genetics (2 out of every 11 mice born is DD), both DD mice and their littermate controls were exposed to multiple treatments. Mice were tested in behavioral experiments until one of the following criteria was reached: (1) the mice had received six drug treatments excluding daily L-dopa injections, (2) the age of the mice reached 10 months, or (3) the mice lost >20% of their body weight or displayed other signs of illness. A total of 40 control and 35 DD mice were used in studies that involved PCP and a total of 37 control and 46 DD mice were used in studies that involved MK-801. On drug testing days, the treatments were randomized, and each mouse was, at some point in the study, treated with either PCP (5 mg/kg) or MK-801 (0.75 mg/kg). Because of our experimental design (mice were continuously added and removed), within-group comparisons using repeated measures were impossible. Therefore, in each figure, comparisons are made between data from mice treated with a drug of interest and pooled data from all mice treated with either vehicle, PCP (5 mg/kg), or MK-801 (0.75 mg/kg).
In Situ Hybridization
In situ hybridization was performed as described (Chartoff et al, 1999, 2001) with minor modifications. Briefly, mice were killed 1 h after treatment with vehicle, PCP, or MK-801, and the brains removed and frozen in isopentane on dry ice. Coronal sections (20 μm) were cut with a cryostat, thaw-mounted on Fisherbrand Superfrost microscope slides, and kept at −80°C until used in the hybridization assay. Tissue sections were fixed and subsequently hybridized with an 35S-UTP-labeled c-fos antisense riboprobe transcribed with SP6 RNA polymerase from a linearized rat c-fos cDNA template. Tissue sections were hybridized overnight at 58°C and washed at 62°C. Final concentration of probe was 1.5 pmol probe/ml hybridization mix. After washing, slides were dehydrated through a graded alcohol series in which water was substituted by 0.6 M ammonium acetate and air-dried. For visualization of the c-fos signal, slides were subsequently dipped in NTB-2 emulsion (Eastman Kodak; diluted 1 : 1 with 0.6 M ammonium acetate), exposed for 4 weeks, developed, and counterstained with cresyl violet. Sense-strand controls for the c-fos riboprobe produced no detectable signal above background (Adams et al, 1997).
Data Analysis
Data assessing pharmacological treatments on locomotor activity in control and DD mice (Figures 1a, b, 2, and 5) were analyzed by two-way ANOVA (genotype × treatment). Behavioral data presented in Figure 4 were analyzed by one-way ANOVA. Any significant main effects and interactions revealed in an ANOVA were further examined using the Bonferroni/Dunn test. Since the Bonferroni/Dunn test corrects for multiple comparisons, significance was set at P<α/n for each experiment, where _α_=0.05 and _n_=number of comparisons. Data from the c-fos in situ hybridization study (Table 1) were analyzed by three-way ANOVA (genotype × treatment × brain region) with repeated measures on brain region, followed by Newman–Keuls post hoc tests.
Figure 1
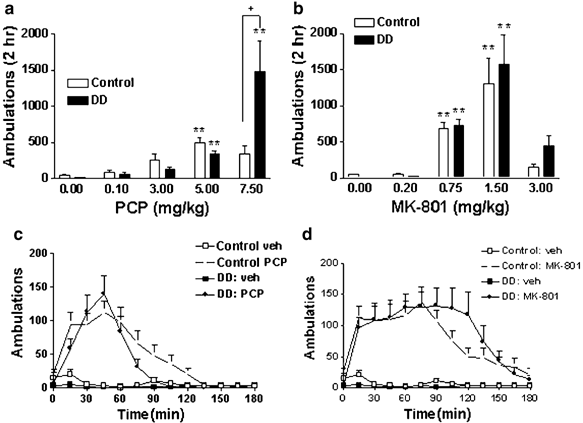
PCP and MK-801 cause hyperlocomotion in both control and DD mice. (a and b) Forward locomotion was measured for 2 h in control and DD mice in response to increasing doses of PCP (a) and MK-801 (b). The number of animals in each group is as follows: PCP vehicle (0.0 mg/kg), control _n_=26, DD _n_=26; PCP 0.1 mg/kg, control _n_=24, DD _n_=16; PCP 3.0 mg/kg, control _n_=16, DD _n_=14; PCP 5.0 mg/kg, control _n_=40, DD _n_=35; PCP 7.5 mg/kg, control _n_=8, DD _n_=8; MK-801 vehicle, control _n_=26, DD _n_=26; MK-801 0.20 mg/kg, control _n_=7, DD _n_=7; MK-801 0.75 mg/kg, control _n_=37, DD _n_=46; MK-801 1.5 mg/kg, control _n_=7, DD _n_=7; MK-801 3.0 mg/kg, control _n_=8, DD _n_=8. **P<0.001 compared to the 0.0 mg/kg dose of the corresponding genotype. +P<0.05 comparing DD and control mice treated with PCP (7.5 mg/kg). (c and d) Time course (0–180 min) of locomotor effects of PCP (5.0 mg/kg) (control n =26, DD _n_=21) (c) and MK-801 (0.75 mg/kg) (control _n_=18, DD _n_=20) (d).
Figure 2
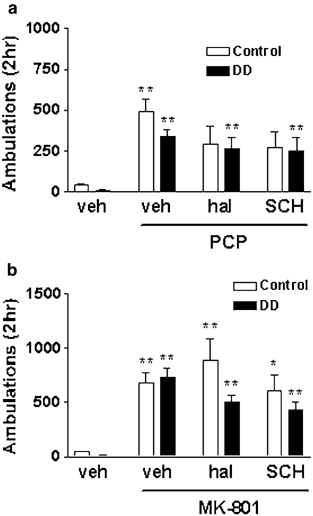
Effect of dopamine receptor antagonists on NMDA receptor antagonist-induced hyperlocomotion. Haloperidol (hal, 0.5 mg/kg) and SCH 23390 (SCH, 0.2 mg/kg) were administered to control or DD mice 15 min prior to PCP (5 mg/kg) (a) or MK-801 (0.75 mg/kg) (b). The number of animals in each group is as follows: vehicle, control _n_=26, DD _n_=26; PCP, control _n_=40, DD _n_=35; hal+PCP, control _n_=13, DD _n_=18; SCH+PCP, control _n_=13, DD _n_=17; MK-801, control _n_=37, DD _n_=46; hal+MK-801, control _n_=22, DD _n_=21; SCH+MK-801, control _n_=22, DD _n_=21. *P<0.008, **P<0.0016 compared to vehicle treatment of the corresponding genotype.
Figure 5
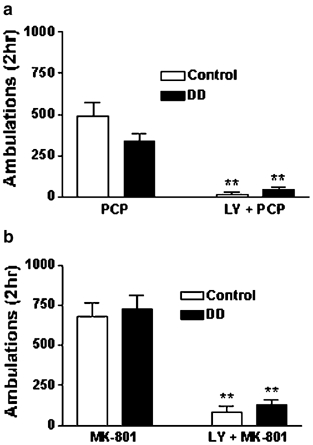
The mGlu 2/3 agonist LY379268 attenuates PCP- and MK-801-induced hyperlocomotion in both control and DD mice. (a) DD and control mice were treated with PCP (5 mg/kg) or LY379268 (LY, 10 mg/kg) plus PCP and locomotor activity was measured for 2 h. (b) DD and control mice were treated with MK-801 (0.75 mg/kg) or LY379268 (LY, 10 mg/kg) plus MK-801 and locomotor activity was measured for 2 h. LY379268 was administered 15 min prior to PCP and MK-801. The number of animals in each group is as follows: PCP, control _n_=40, DD _n_=35; LY+PCP, control _n_=8, DD _n_=9; MK-801, control _n_=37, DD _n_=46; LY+MK-801, control _n_=8, DD _n_=8. **P<0.01 compared to PCP or MK-801 alone of the corresponding genotype.
Figure 4
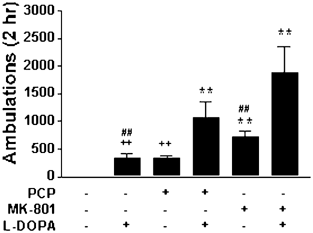
L-Dopa potentiates NMDA receptor antagonist-induced hyperlocomotion. DD mice were treated with vehicle (_n_=26), L-dopa (20 mg/kg) (_n_=20), PCP (5 mg/kg) (_n_=35), MK-801 (0.75 mg/kg) (_n_=46), PCP and L-dopa (_n_=12), or MK-801 and L-dopa (_n_=7) and locomotor activity was measured for 2 h. PCP and MK-801 were administered 15 min prior to L-dopa. **P<0.0006 compared to vehicle;++P<0.0006 compared to PCP and L-dopa; ##P<0.0006 compared to MK-801 and L-dopa.
Table 1 Quantification of c_-fos_-Positive Cells
RESULTS
PCP and MK-801 Stimulate Locomotion in Both Control and DD Mice
To test whether dopamine is required for the locomotor response to NMDA receptor antagonists, both control and DD animals were injected with PCP (0. 0, 0.1, 3.0, 5.0, and 7.5 mg/kg) or MK-801 (0.0, 0.2, 0.75, 1.5, and 3.0 mg/kg) and locomotor activity was measured for 2 h. The effects of PCP on locomotor activity were genotype- and dose-dependent (genotype × dose interaction: F(4,203)=11.79, P<0.0001) (Figure 1a). Compared to vehicle-treated mice of the corresponding genotype, 5.0 mg/kg PCP significantly increased locomotion in both control and DD mice. Locomotor activity in DD mice treated with PCP (7.5 mg/kg) was significantly greater than that of control mice treated with PCP (7.5 mg/kg). Control mice treated with 7.5 mg/kg PCP engaged primarily in stereotyped behaviors, such as grooming and rearing as has been documented previously (Sturgeon et al, 1979), while DD mice exhibited primarily locomotor activity. A higher dose of PCP (10.0 mg/kg) resulted in a stumbling gait and immobility and was therefore not tested further (data not shown). There was no statistically significant difference between control and DD mice at a dose of 3.0 mg/kg PCP. The effects of MK-801 on locomotor activity were dose-dependent (dose: F(4,169)=31.83, P<0.0001): 0.75 and 1.5 mg/kg MK-801 significantly increased locomotion in both control and DD mice compared to vehicle-treated mice of the corresponding genotype (Figure 1b). At 3.0 mg/kg MK-801, locomotor activity appeared uncoordinated and mice had difficulty changing direction in the corners of the chambers as has been noted previously (Carlsson and Carlsson, 1989b). There were no statistical differences between genotypes within each dose of MK-801. The time course of PCP (5 mg/kg)- and MK-801 (0.75 mg/kg)-induced locomotor activity was qualitatively similar between control and DD mice (Figure 1c and d).
Effect of Dopamine Receptor Antagonists on PCP- and MK-801-Induced Hyperlocomotion
To confirm that the hyperlocomotor response of the DD mice to either PCP or MK-801 was not due to signaling through dopamine receptors, we administered either the dopamine D2 receptor antagonist haloperidol or the dopamine D1 receptor antagonist SCH 23390 to control and DD mice 15 min prior to treatment with NMDA receptor antagonists. The dose of haloperidol chosen (0.5 mg/kg) has been shown to block amphetamine-induced locomotion but not to induce cataleptic behavior in mice (Heusner et al, 2003). The dose of SCH 23390 chosen has been shown to block dopamine-induced behaviors and c_-fos_ induction in DD mice (Chartoff et al, 2001). Locomotor activity in control and DD mice treated with PCP (5.0 mg/kg) was significantly increased compared to vehicle treatment (treatment: F(3,180)=16.92, P<0.0001) (Figure 2a). However, locomotor activity in control mice treated with either haloperidol (0.5 mg/kg) or SCH 23390 (0.2 mg/kg) prior to PCP was not significantly increased compared to vehicle. In contrast, locomotor activity in DD mice treated with haloperidol and PCP, or SCH 23390 and PCP was significantly increased compared to vehicle. Pretreatment of either control or DD mice with the dopamine receptor antagonists did not significantly attenuate PCP-induced locomotion. Locomotor activity in control and DD mice treated with MK-801 (0.75 mg/kg), haloperidol (0.5 mg/kg) and MK-801 (0.75 mg/kg), or SCH 23390 (0.2 mg/kg) and MK-801 was significantly increased compared to vehicle treatment of the corresponding genotype (treatment: F(3,213)=20.02, P<0.0001) (Figure 2b). Pretreatment of either control or DD mice with the dopamine receptor antagonists did not significantly attenuate MK-801-induced locomotion.
PCP and MK-801 Induce Striatal c-fos Expression
The immediate-early gene c-fos is an indicator of transcriptional activity, and PCP has previously been shown to increase striatal expression of c_-fos_ in wild-type mice (Svenningsson et al, 2003). The striatum receives extensive dopaminergic input and is critical for the regulation of movement (Ungerstedt, 1971; Graybiel et al, 1990). In situ hybridization was used to assess c_-fos_ mRNA in the striatum of control and DD mice. A 1-h treatment with PCP at 5.0 mg/kg (Figure 3e–g) or MK-801 at 0.75 mg/kg (Figure 3i–l) induced c-fos mRNA in scattered cells of the dorsomedial striatum (DMSt) and NAc in both control and DD mice. The c-fos_-positive cells in these brain regions were distinct and heavily labeled compared to vehicle-treated control sections (Figure 3a–d) that showed faint and sparse background levels of c-fos expression. The number of c_-fos_-positive cells was quantified in a subset of sections (see Table 1). Both PCP and MK-801 significantly increased c-fos in the two brain regions (treatment × region interaction: F(2,12)=4.22, P<0.05). There was no difference between genotypes within treatments or brain regions: (genotype × treatment: F(2,12)=0.48, NS) and (genotype × region: F(1,12)=0.85, NS). Induction of c_-fos by PCP and MK-801 was limited to the DMSt and NAc and was largely absent from the dorsolateral striatum.
Figure 3
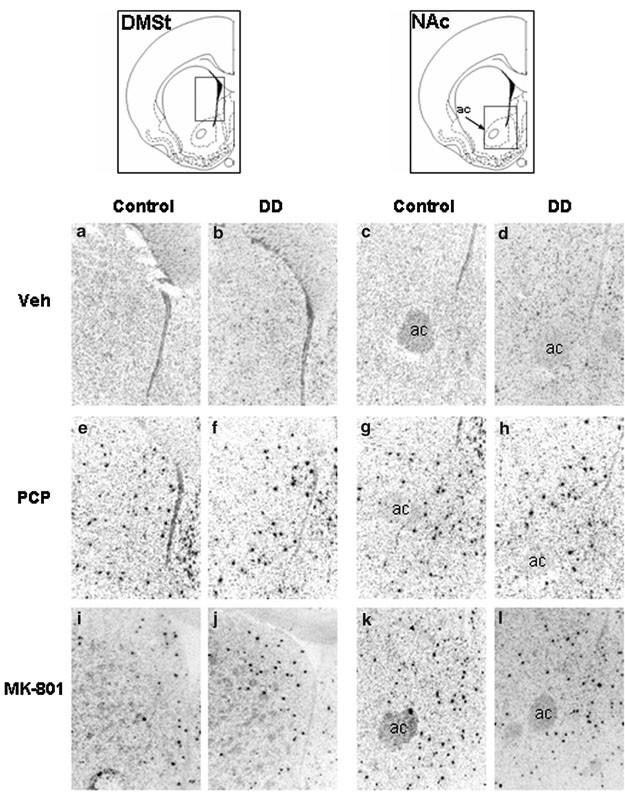
PCP and MK-801 induce c-fos mRNA expression in the striatum and NAc of control and DD mice. Mice were injected with vehicle (veh), PCP (5 mg/kg), or MK-801 (0.75 mg/kg) and killed for in situ hybridization 60 min later. (a and c) Control mice, vehicle treatment, DMSt and NAc, respectively. (b and d) DD mice, vehicle treatment, DMSt and NAc. (e and g) Control mice, PCP treatment, DMSt and NAc. (f and h) DD mice, PCP treatment, DMSt and NAc. (i and k) Control mice, MK-801 treatment, DMSt and NAc. (j and l) DD mice, MK-801 treatment, DMSt and NAc. A similar induction of c-_fos_-positive cells (black) is visible for each treatment, regardless of genotype. Representative sections for each treatment and genotype are shown.
NMDA Receptor Antagonists and L-Dopa Have an Additive Effect on Locomotor Activity in DD Mice
To test whether dopamine can modulate NMDA receptor antagonist-induced locomotor activity, a low dose of L-dopa (20 mg/kg) was administered to DD mice 15 min after either PCP or MK-801 and ambulations were recorded for 2 h. This dose of L-dopa has previously been shown to induce approximately 20% of the hyperlocomotor response observed with the daily dose of L-dopa (50 mg/kg) in DD mice (Szczypka et al, 1999). We chose this threshold dose because it does not maximize locomotor activity in DD mice and would allow the detection of potentiating effects of dopamine on the behaviors induced by NMDA receptor antagonists. There was a significant effect of treatment on locomotor activity (F(5,140)=18.98, P<0.0001) (Figure 4). L-Dopa (20 mg/kg) increased locomotor activity to a level qualitatively comparable to that of PCP (5.0 mg/kg), but neither drug caused statistically significant increases in locomotion compared to vehicle using the Bonferroni/Dunn test for multiple comparisons. Administration of L-dopa to PCP-treated mice increased the number of ambulations by a factor of 3. Administration of L-dopa to MK-801-treated (0.75 mg/kg) mice increased the number of ambulations slightly more than two-fold.
An mGlu 2/3 Agonist Attenuates PCP- and MK-801-Induced Locomotion in Both Control and DD Mice
To determine whether glutamate release is required for NMDA antagonist-induced locomotion in DD mice, we administered LY379268 (10 mg/kg) 15 min prior to either PCP (5.0 mg/kg) or MK-801 (0.75 mg/kg) and recorded locomotor activity for 2 h. Treatment with LY379268 blocked both PCP- (treatment: F(1,88)=15.75, P<0.0001) and MK-801- (treatment: F(1,95)=16.74, P<0.0001) induced locomotion in control and DD mice (Figure 5a and b). LY379268 alone did not significantly affect basal locomotor activity (data not shown).
DISCUSSION
This study uses mice with a genetic deletion of tyrosine hydroxylase specifically in dopaminergic neurons to demonstrate that dopamine is not required for behavioral or transcriptional responses to PCP or MK-801. Furthermore, we provide evidence that glutamatergic neurotransmission is necessary for NMDA receptor antagonist-induced locomotor activity. Specifically, both PCP and MK-801 induced locomotor activity to a similar extent in control and DD mice. Also, neither haloperidol nor SCH 23390 significantly attenuated PCP- or MK-801-induced hyperlocomotion in either control or DD mice. Furthermore, PCP and MK-801 induced similar levels and expression patterns of c_-fos_ mRNA in the DMSt and NAc of both control and DD mice. Finally, PCP- and MK-801-induced locomotion was blocked by LY379268, an mGlu 2/3 agonist that acts presynaptically to inhibit glutamate release.
Our data are consistent with studies that suggest that dopamine is not required for the effects of NMDA receptor antagonists. For example, NMDA receptor antagonists have been shown to increase locomotor activity in animals with temporary catecholamine depletion (Carlsson and Carlsson, 1989a; Lapin and Rogawski, 1995; Swanson and Schoepp, 2002). Also, the reinforcing and locomotor stimulatory effects of PCP and MK-801 have been dissociated from dopamine (Carlezon and Wise, 1996; Druhan et al, 1996; Pierce et al, 1997; Adams and Moghaddam, 1998). However, our findings contrast with some studies that demonstrate a requirement for dopamine in mediating the effects of NMDA receptor antagonists. For example, dopamine receptor antagonists have been shown to reduce the locomotor stimulatory effects of PCP and MK-801 (Sturgeon et al, 1981; Freed et al, 1984; Lapin and Rogawski, 1995). Also, 6-OHDA lesions can block NMDA receptor antagonist-induced behaviors (French and Vantini, 1984; French et al, 1985; Steinpreis and Salamone, 1993).
The discrepancies between the present data and previous studies with antagonists and pharmacological lesions may be explained by several inherent disadvantages of these methods. For example, 6-OHDA lesions are toxic to cells, and the percentage of dopaminergic neurons lesioned is variable and does not usually exceed 95%. Also, pharmacological agents are often nonspecific and affect neurotransmitter systems other than dopamine. In the context of these methodological issues, the variability in results found in many studies is understandable. A potential limitation of using DD mice is that neuroadaptive changes may occur throughout development to compensate for the chronic lack of tyrosine hydroxylase in dopaminergic neurons. Although DD mice lack dopamine from embryogenesis onward, there are no differences in the levels of dopamine receptors or the dopamine transporter compared to control mice (Kim et al, 2000). The basic electrophysiological properties of dopaminergic neurons are also normal (Robinson et al, 2004). Likewise, light and electron microscopic analysis of midbrain dopaminergic neurons, their projections, and target neurons in the striatum of DD mice has not revealed any gross differences compared to control mice (Zhou and Palmiter, 1995; Kim et al, 2002). In addition, the glutamatergic input to the striatum appears to be normal in that glutamate labeling in synaptic vesicles is normal, extracellular glutamate is normal as measured by microdialysis, and the kinetics of synaptic vesicle release from corticostriatal projections is normal in slice preparations (Bamford et al, 2004). The only difference noted thus far is that signaling via either D1 or D2 receptors is greatly enhanced in DD mice compared to controls, such that lower levels of dopamine or dopamine receptor agonists elicit maximal molecular or behavioral effects in DD mice (Kim et al, 2000, 2002; Robinson et al, 2004). In fact, restoration of dopamine to normal levels in DD mice results in intense stereotypy (Chartoff et al, 2001). This hypersensitivity phenomenon is similar to that observed in animals lesioned with 6-OHDA (Zigmond and Stricker, 1984) and can be reversed in DD mice by chronic treatment with L-dopa (Kim et al, 2000). While it is impossible to conclude that DD mice are identical to control mice except for the lack of dopamine (and the hypersensitivity to dopamine signaling when dopamine is restored), the existing evidence suggests that DD mice provide a useful model of dopamine depletion that is not encumbered by other neuroadaptations.
In the present study, the locomotor response of DD and control mice to PCP and MK-801 was similar but not identical. Notably, the highest dose of PCP (7.5 mg/kg) elicited significantly more ambulations in DD than in control mice, and there was a similar trend for the highest dose of MK-801 (3.0 mg/kg). There is evidence for a behavioral supersensitivity to NMDA receptor antagonists in rats with neonatal 6-OHDA lesions of the NAc (Moy and Breese, 2002). Likewise, we observed that the control mice were less ambulatory because they were engaged in more stereotypic behaviors. Stereotypy and locomotion are thought to be competing motor activities that involve distinct mechanisms (Segal and Mandell, 1974), and increased striatal dopamine D1 receptor activation may be responsible for the emergence of stereotypies in control mice (Chartoff et al, 2001).
While we have used PCP and MK-801 to determine the role of dopamine in the locomotor response to NMDA receptor antagonists, these compounds are not identical. This is revealed in both the dose–response curve, where the response of DD and control mice to PCP is more disparate than the responses to MK-801, and in the fact that PCP-induced locomotion in control mice is more affected by dopamine receptor antagonists than MK-801-induced locomotion in control mice. MK-801 can elicit locomotor activity at doses that do not cause detectable increases in extracellular dopamine (Druhan et al, 1996), and PCP, but not MK-801, can act as a dopamine uptake inhibitor (Gerhardt et al, 1987). Therefore, it may be that dopamine has more of a facilitatory role with PCP than with MK-801 in control mice.
The mechanisms responsible for NMDA receptor antagonist-induced locomotion and c-fos expression in the absence of dopamine are currently unknown. One possibility is that they lead to an increase in the activation of glutamatergic transmission at non-NMDA receptors, which in turn drives behavior and transcriptional responses. NMDA receptor antagonists have been shown to increase extracellular glutamate levels in both the prefrontal cortex and NAc (Adams and Moghaddam, 1998; Moghaddam and Adams, 1998). It has been proposed that this increase in glutamate may be due to an inhibition of tonic GABA inputs onto cortical pyramidal neurons (Grunze et al, 1996; Takahata and Moghaddam, 2003). The prefrontal cortex projects to the DMSt and NAc (Sesack et al, 1989), and increased glutamate release into these regions could stimulate non-NMDA receptors, causing cellular activation and locomotion. In such a model for behavioral activation by NMDA receptor antagonists, the release of dopamine is not required for evoking a locomotor response.
To test whether increased glutamate release was responsible for the locomotor activating effects of PCP and MK-801 in DD and control mice, we utilized the mGlu 2/3 agonist LY379268. mGlu 2/3 receptors are localized to corticostriatal axon terminals that synapse in the ventral striatum, and their activation causes long-term depression of excitatory synaptic transmission (Robbe et al, 2002). These receptors have also been localized to glial processes, but are absent from postsynaptic dendrites and neuronal cell bodies in the striatum (Jokel et al, 2001; Robbe et al, 2002). We found that LY379268 blocked PCP- and MK-801-induced locomotion in both control and DD mice. This is consistent with previous work showing that treatment of intact and monoamine-depleted rats with the mGlu 2/3 agonist LY379268 inhibits PCP-induced hyperlocomotion (Moghaddam and Adams, 1998; Swanson and Schoepp, 2002). Antagonists of mGlu 2/3 receptors have been shown to increase locomotor activity in mice (O'Neill et al, 2003). The effect of cotreatment of mGlu 2/3 antagonists and PCP is variable: one group found an increase in PCP-induced motor activity (Olszewski et al, 2004), yet another group found an incomplete effect on PCP self-administration (Winter et al, 2004).
Our results do not explicitly address what neural circuits are involved in the response of control and DD mice to NMDA receptor antagonists. A parsimonious suggestion is that the same corticostriatal circuits that respond to enhanced extracellular dopamine (eg in response to cocaine or amphetamine) can also respond to enhanced extracellular glutamate. However, we cannot rule out the possibility that glutamatergic activation of neural circuits either upstream or downstream of the corticostriatal input is also required for the locomotor response to NMDA receptor antagonists.
Further support for the suggestion that dopamine-responsive corticostriatal circuitry is critical for mediating the effects of NMDA receptor antagonists is our finding that temporary and partial restoration of dopamine synthesis by L-dopa treatment greatly potentiated PCP- and MK-801-induced hyperlocomotion. It has previously been shown by Greenberg and Segal (1985) that repeated daily injections of amphetamine sensitize rats to the locomotor-stimulating effects of PCP. These authors suggest that the enhanced locomotor activity may be due to an increase in the PCP-stimulated release of mesolimbic dopamine. This is consistent with the idea that dopamine is not required for, but can contribute to, NMDA receptor antagonist-induced hyperlocomotion. It is unlikely that PCP- and MK-801-induced locomotion in DD mice that have not been treated with L-dopa for 18 h is due to a similar increase in dopamine because dopamine levels at this time point are barely detectable at <1% of control.
While glutamate and dopamine may normally act in concert within the striatum, particularly the NAc, to stimulate locomotion, our data suggest that enhanced glutamatergic signaling in corticostriatal circuitry in response to PCP and MK-801 may be sufficient to stimulate behaviors such as locomotion. This is consistent with previous findings (Adams and Moghaddam, 1998; Takahata and Moghaddam, 2003) that showed a temporal dissociation between dopamine release and the locomotor response to PCP and suggests that dopamine release may be an indirect effect of activated glutamate release. As such, the efficacy of dopamine receptor antagonists at blocking NMDA receptor antagonist-induced locomotion can be explained by the hypothesis that dopamine activation is downstream of glutamatergic activation of critical pathways, and blockade of dopamine receptors will impede activity. However, as dopamine receptor antagonists often have undesirable side effects, it appears more effective to target the critical glutamatergic signaling to inhibit behaviors induced by NMDA receptor antagonists. If NMDA receptor antagonist-induced hyperlocomotion is relevant to aspects of psychosis in humans, then our data along with others (Moghaddam and Adams, 1998; Swanson and Schoepp, 2002) underscore the importance of modulating glutamatergic, rather than dopaminergic, signaling when studying schizophrenia and related disorders in humans.
References
- Adams B, Moghaddam B (1998). Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci 18: 5545–5554.
Article CAS Google Scholar - Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS (1997). Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci USA 94: 12157–12161.
Article CAS Google Scholar - Andine P, Widermark N, Axelsson R, Nyberg G, Olofsson U, Martensson E et al (1999). Characterization of MK-801-induced behavior as a putative rat model of psychosis. J Pharmacol Exp Ther 290: 1393–1408.
CAS PubMed Google Scholar - Bakshi VP, Swerdlow NR, Geyer MA (1994). Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther 271: 787–794.
CAS Google Scholar - Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK (2004). Dopamine modulates release from corticostriatal terminals. J Neurosci 24: 9541–9552.
Article CAS Google Scholar - Carlezon Jr WA, Wise RA (1996). Rewarding actions of phencyclidine and related drugs in nucleus accumbens shell and frontal cortex. J Neurosci 16: 3112–3122.
Article CAS Google Scholar - Carlsson M, Carlsson A (1989a). The NMDA antagonist MK-801 causes marked locomotor stimulation in monoamine-depleted mice. J Neural Transm 75: 221–226.
Article CAS Google Scholar - Carlsson M, Carlsson A (1989b). Dramatic synergism between MK-801 and clonidine with respect to locomotor stimulatory effect in monoamine-depleted mice. J Neural Transm 77: 65–71.
Article CAS Google Scholar - Chartoff EH, Marck BT, Matsumoto AM, Dorsa DM, Palmiter RD (2001). Induction of stereotypy in dopamine-deficient mice requires striatal D1 receptor activation. Proc Natl Acad Sci USA 98: 10451–10456.
Article CAS Google Scholar - Chartoff EH, Ward RP, Dorsa DM (1999). Role of adenosine and _N_-methyl-D-aspartate receptors in mediating haloperidol-induced gene expression and catalepsy. J Pharmacol Exp Ther 291: 531–537.
CAS PubMed Google Scholar - Cornish JL, Nakamura M, Kalivas PW (2001). Dopamine-independent locomotion following blockade of _N_-methyl-D-aspartate receptors in the ventral tegmental area. J Pharmacol Exp Ther 298: 226–233.
CAS PubMed Google Scholar - Coyle JT (1996). The glutamatergic dysfunction hypothesis for schizophrenia. Harvard Rev Psychiatry 3: 241–253.
Article CAS Google Scholar - Creese I, Iversen SD (1973). Blockage of amphetamine induced motor stimulation and stereotypy in the adult rat following neonatal treatment with 6-hydroxydopamine. Brain Res 55: 369–382.
Article CAS Google Scholar - Davis KL, Kahn RS, Ko G, Davidson M (1991). Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry 148: 1474–1486.
Article CAS Google Scholar - Druhan JP, Rajabi H, Stewart J (1996). MK-801 increases locomotor activity without elevating extracellular dopamine levels in the nucleus accumbens. Synapse 24: 135–146.
Article CAS Google Scholar - Freed WJ, Bing LA, Wyatt RJ (1984). Effects of neuroleptics on phencyclidine (PCP)-induced locomotor stimulation in mice. Neuropharmacology 23: 175–181.
Article CAS Google Scholar - Freeman AS, Bunney BS (1984). The effects of phencyclidine and _N_-allylnormetazocine on midbrain dopamine neuronal activity. Eur J Pharmacol 104: 287–293.
Article CAS Google Scholar - French ED, Mura A, Wang T (1993). MK-801, phencyclidine (PCP), and PCP-like drugs increase burst firing in rat A10 dopamine neurons: comparison to competitive NMDA antagonists. Synapse 13: 108–116.
Article CAS Google Scholar - French ED, Pilapil C, Quirion R (1985). Phencyclidine binding sites in the nucleus accumbens and phencyclidine-induced hyperactivity are decreased following lesions of the mesolimbic dopamine system. Eur J Pharmacol 116: 1–9.
Article CAS Google Scholar - French ED, Vantini G (1984). Phencyclidine-induced locomotor activity in the rat is blocked by 6-hydroxydopamine lesion of the nucleus accumbens: comparisons to other psychomotor stimulants. Psychopharmacology (Berl) 82: 83–88.
Article CAS Google Scholar - Gerhardt GA, Pang K, Rose GM (1987). In vivo electrochemical demonstration of the presynaptic actions of phencyclidine in rat caudate nucleus. J Pharmacol Exp Ther 241: 714–721.
CAS PubMed Google Scholar - Graybiel AM, Hirsch EC, Agid Y (1990). The nigrostriatal system in Parkinson's disease. Adv Neurol 53: 17–29.
CAS PubMed Google Scholar - Greenberg BD, Segal DS (1985). Acute and chronic behavioral interactions between phencyclidine (PCP) and amphetamine: evidence for a dopaminergic role in some PCP-induced behaviors. Pharmacol Biochem Behav 23: 99–105.
Article CAS Google Scholar - Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW et al (1996). NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16: 2034–2043.
Article CAS Google Scholar - Heusner CL, Hnasko TS, Szczypka MS, Liu Y, During MJ, Palmiter RD (2003). Viral restoration of dopamine to the nucleus accumbens is sufficient to induce a locomotor response to amphetamine. Brain Res 980: 266–274.
Article CAS Google Scholar - Imperato A, Scrocco MG, Bacchi S, Angelucci L (1990). NMDA receptors and in vivo dopamine release in the nucleus accumbens and caudatus. Eur J Pharmacol 187: 555–556.
Article CAS Google Scholar - Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Article CAS Google Scholar - Jokel ES, Garduno ER, Ariano MA, Levine MS (2001). Metabotropic glutamate receptors mGluR1alpha and mGluR2/3 display dynamic expression patterns in developing rat striatum. Dev Neurosci 23: 1–6.
Article CAS Google Scholar - Kelly PH, Seviour PW, Iversen SD (1975). Amphetamine and apomorphine responses in the rat following 6-OHDA lesions of the nucleus accumbens septi and corpus striatum. Brain Res 94: 507–522.
Article CAS Google Scholar - Kim DS, Froelick GJ, Palmiter RD (2002). Dopamine-dependent desensitization of dopaminergic signaling in the developing mouse striatum. J Neurosci 22: 9841–9849.
Article CAS Google Scholar - Kim DS, Szczypka MS, Palmiter RD (2000). Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci 20: 4405–4413.
Article CAS Google Scholar - Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmuller B (1980). Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett 20: 379–382.
Article CAS Google Scholar - Lapin IP, Rogawski MA (1995). Effects of D1 and D2 dopamine receptor antagonists and catecholamine depleting agents on the locomotor stimulation induced by dizocilpine in mice. Behav Brain Res 70: 145–151.
Article CAS Google Scholar - Miller DW, Abercrombie ED (1996). Effects of MK-801 on spontaneous and amphetamine-stimulated dopamine release in striatum measured with in vivo microdialysis in awake rats. Brain Res Bull 40: 57–62.
Article CAS Google Scholar - Moghaddam B, Adams BW (1998). Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281: 1349–1352.
Article CAS Google Scholar - Moy SS, Breese GR (2002). Phencyclidine supersensitivity in rats with neonatal dopamine loss. Psychopharmacology (Berl) 161: 255–262.
Article CAS Google Scholar - O'Neill MF, Heron-Maxwell C, Conway MW, Monn JA, Ornstein P (2003). Group II metabotropic glutamate receptor antagonists LY341495 and LY366457 increase locomotor activity in mice. Neuropharmacology 45: 565–574.
Article CAS Google Scholar - Olney JW, Farber NB (1995). Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52: 998–1007.
Article CAS Google Scholar - Olszewski RT, Bukhari N, Zhou J, Kozikowski AP, Wroblewski JT, Shamimi-Noori S et al (2004). NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J Neurochem 89: 876–885.
Article CAS Google Scholar - Pierce RC, Meil WM, Kalivas PW (1997). The NMDA antagonist, dizocilpine, enhances cocaine reinforcement without influencing mesoaccumbens dopamine transmission. Psychopharmacology (Berl) 133: 188–195.
Article CAS Google Scholar - Rios M, Habecker B, Sasaoka T, Eisenhofer G, Tian H, Landis S et al (1999). Catecholamine synthesis is mediated by tyrosinase in the absence of tyrosine hydroxylase. J Neurosci 19: 3519–3526.
Article CAS Google Scholar - Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni OJ (2002). Role of p/q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci 22: 4346–4356.
Article CAS Google Scholar - Robinson S, Smith DM, Mizumori SJ, Palmiter RD (2004). Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci USA 101: 13329–13334.
Article CAS Google Scholar - Segal DS, Mandell AJ (1974). Long-term administration of _d_-amphetamine: progressive augmentation of motor activity and stereotypy. Pharmacol Biochem Behav 2: 249–255.
Article CAS Google Scholar - Sesack SR, Deutch AY, Roth RH, Bunney BS (1989). Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol 290: 213–242.
Article CAS Google Scholar - Snyder SH (1976). The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry 133: 197–202.
Article CAS Google Scholar - Steinpreis RE, Salamone JD (1993). The role of nucleus accumbens dopamine in the neurochemical and behavioral effects of phencyclidine: a microdialysis and behavioral study. Brain Res 612: 263–270.
Article CAS Google Scholar - Sturgeon RD, Fessler RG, London SF, Meltzer HY (1981). A comparison of the effects of neuroleptics on phencyclidine-induced behaviors in the rat. Eur J Pharmacol 76: 37–53.
Article CAS Google Scholar - Sturgeon RD, Fessler RG, Meltzer HY (1979). Behavioral rating scales for assessing phencyclidine-induced locomotor activity, stereotyped behavior and ataxia in rats. Eur J Pharmacol 59: 169–179.
Article CAS Google Scholar - Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M et al (2003). Diverse psychotomimetics act through a common signaling pathway. Science 302: 1412–1415.
Article CAS Google Scholar - Swanson CJ, Schoepp DD (2002). The group II metabotropic glutamate receptor agonist (−)-2-oxa-4-aminobicyclo[3.1.0.]hexane-4, 6-dicarboxylate (LY379268) and clozapine reverse phencyclidine-induced behaviors in monoamine-depleted rats. J Pharmacol Exp Ther 303: 919–927.
Article CAS Google Scholar - Szczypka MS, Rainey MA, Kim DS, Alaynick WA, Marck BT, Matsumoto AM et al (1999). Feeding behavior in dopamine-deficient mice. Proc Natl Acad Sci USA 96: 12138–12143.
Article CAS Google Scholar - Takahata R, Moghaddam B (2003). Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology 28: 1117–1124.
Article CAS Google Scholar - Thomas SA, Marck BT, Palmiter RD, Matsumoto AM (1998). Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem 70: 2468–2476.
Article CAS Google Scholar - Ungerstedt U (1971). Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiol Scand Suppl 367: 95–122.
Article CAS Google Scholar - Ungerstedt U, Ljungberg T, Steg G (1974). Behavioral, physiological, and neurochemical changes after 6-hydroxydopamine-induced degeneration of the nigro-striatal dopamine neurons. Adv Neurol 5: 421–426.
CAS PubMed Google Scholar - Verma A, Moghaddam B (1996). NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci 16: 373–379.
Article CAS Google Scholar - Winter JC, Eckler JR, Rabin RA (2004). Serotonergic/glutamatergic interactions: the effects of mGlu2/3 receptor ligands in rats trained with LSD and PCP as discriminative stimuli. Psychopharmacology (Berl) 172: 233–240.
Article CAS Google Scholar - Zhou QY, Palmiter RD (1995). Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 83: 1197–1209.
Article CAS Google Scholar - Zigmond MJ, Stricker EM (1984). Parkinson's disease: studies with an animal model. Life Sci 35: 5–18.
Article CAS Google Scholar
Acknowledgements
We thank Bethany Sotak and Nora Meneses for DD colony maintenance, Victor Denenberg for advice on statistical analyses, Anne Marie Wissman and Eliot Brenowitz for use of their microscope, and Bill Carlezon for the use of his laboratory facilities at McLean Hospital, Belmont, MA to conduct in situ hybridization experiments. This study was supported by USPHS Grant T32-DA07278 (to CLH) from NIDA.
Author information
Author notes
- Elena H Chartoff and Carrie L Heusner: These authors contributed equally to this work
Authors and Affiliations
- Department of Psychiatry, Harvard Medical School, McLean Hospital Belmont, MA, USA
Elena H Chartoff - Department of Biochemistry, Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA
Carrie L Heusner & Richard D Palmiter - Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA
Richard D Palmiter
Authors
- Elena H Chartoff
You can also search for this author inPubMed Google Scholar - Carrie L Heusner
You can also search for this author inPubMed Google Scholar - Richard D Palmiter
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRichard D Palmiter.
Rights and permissions
About this article
Cite this article
Chartoff, E., Heusner, C. & Palmiter, R. Dopamine is not Required for the Hyperlocomotor Response to NMDA Receptor Antagonists.Neuropsychopharmacol 30, 1324–1333 (2005). https://doi.org/10.1038/sj.npp.1300678
- Received: 29 June 2004
- Revised: 03 December 2004
- Accepted: 06 December 2004
- Published: 26 January 2005
- Issue Date: 01 July 2005
- DOI: https://doi.org/10.1038/sj.npp.1300678