Methylation in hMLH1 promoter interferes with its binding to transcription factor CBF and inhibits gene expression (original) (raw)
Microsatellite instability (MSI) has been shown to be involved in a pathway of colorectal cancer development distinct from the other genetic pathway including alterations in APC/β-catenin, K-ras and p53 (Aaltonen et al., 1993; Thibodeau et al., 1993; Kinzler and Vogelstein, 1996). MSI is caused by the dysfunction of mismatch repair genes (Papadopoulos et al., 1994). Loss of expression of mismatch repair gene hMLH1 is frequently observed in sporadic colon cancer with MSI (Thibodeau et al., 1996). Methylation of CpG sites in the promoter region of hMLH1 has been shown to correlate with the loss of gene expression (Kane et al., 1997; Cunningham et al., 1998; Herman et al., 1998; Veigl et al., 1998; Deng et al., 1999). This observation along with other correlation between the methylation and the silencing of APC (Hiltunen et al., 1997), BRCA1 (Dobrovic and Simpfendorfer, 1997), p16INK4a (Herman et al., 1995), Rb (Stirzaker et al., 1997), VHL (Herman et al., 1994), E-cadherin (Kanai et al., 1997), estrogen receptor (Issa et al., 1994) genes revealed that DNA methylation is a potent silencer of gene expression. Thus, it is of importance to study the mechanisms of hMLH1 gene silencing by methylation to further elucidate the epigenetic mechanisms of the silencing of mismatch repair genes in the development and progression of cancers.
From our previous work (Deng et al., 1999), a proximal region of hMLH1 promoter (from −248 to −178, 1 indicates the transcription start site) has been identified, in which methylation invariably correlates with silencing of the gene (Figure 1a). To investigate the functional significance of the promoter region of hMLH1 gene, luciferase reporter constructs carrying various 5′ genomic sequences of hMLH1 promoter were constructed as follows: Upstream primers which contain the hMLH1 promoter sequences from −1045 to −1030, from −734 to −717, from −575 to −560, from −258 to −242, from −175 to −159, from −107 to −92 and downstream primer which contains the sequence from +4 to +19 were used to perform polymerase chain reaction (PCR). To yield restriction ends for ligation, sequences 5′-GCGGTACC-3′ and 5′-GCAGATCT-3′ were added to the 5′ ends of the upstream and downstream primers, respectively. The PCR products were digested with restriction enzymes _Kpn_I and _Bgl_II, and ligated to the _Kpn_I and _Bgl_II digested plasmid pGL3-Basic (Promega, Madison, WI, USA), which carries firefly luciferase gene. The plasmid constructs were named as pGL3-1045, pGL3-734, pGL3-575, pGL3-258, pGL3-175 and pGL3-107, respectively. Two other constructs, which carry the promoter sequence (from −272 to +19) with wild type CCAAT box and mutant CCAAT box replaced with TTGGC were obtained by the similar procedure, and named as pGL3-W and pGL3-M respectively.
Figure 1

Reduction of luciferase activities of cells transfected with methylated plasmids. (a) hMLH1 promoter was arbitrarily divided into four regions (A–D). Numbers above the promoter represent the positions of the borders, using 1 as the transcription start site (indicated by an arrow). The numbers of CpG sites, and the average percentages of methylation at all CpG sites in different regions are shown under the promoter (Deng et al., 1999). (b) RKO cells were transfected with _Hha_I methylase-treated plasmids pGL3-107, pGL3-258 and pGL3-575. The luciferase activities of these transfectants (grey bars) were compared respectively with the transfectants from the untreated plasmids (open bars). The open and filled circles represent the unmethylated and methylated CpG sites respectively. (c) Same as in b except that the plasmids were methylated with _Hpa_II methylase
Plasmids pGL3-1045, pGL3-734, pGL3-575, pGL3-258, pGL3-175, pGL3-107, pGL3-Basic and pGL3-Promoter which carries SV40 promoter upstream of the luciferase gene (Promega) were used to transfect a colorectal cancer cell line HT29, which expresses hMLH1 gene. HT29 cells were seeded at 2×105 cells per well in a 24-well plate, and grown in DMEM supplemented with 10% fetal bovine serum. After 24 h, cells were transfected with plasmid constructs using transfection reagent Tfx-50 (Promega) according to the protocol recommended by the manufacturers. A plasmid pRL which carries Renilla luciferase gene was co-transfected with the constructs containing firefly luciferase gene (pGL3) in all the assays mentioned above. Two days after the transfection, cells were lysed, and the luciferase activities were assayed by Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's procedure, and measured with Luminometer (Monolight 2010, Analytical Luminescence Lab., San Diego, CA, USA). The luciferase level was shown by the ratio of firefly luciferase reading over the Renilla luciferase reading. The relative luciferase activity for each transfectant was expressed by its luciferase level divided by that from pGL3-Promoter transfectant.
The luciferase activities obtained from different plasmids are summarized in Figure 2a. Cells transfected with pGL3-107 showed significant luciferase activity compared with pGL3-Basic transfectant. When cells were transfected with plasmids containing longer promoters (e.g. pGL3-175 and pGL3-258), the luciferase activities increased dramatically. However, cells transfected with even longer promoters than pGL3-258 did not show higher luciferase activities (e.g. pGL3-575, pGL3-734 and pGL3-1045). This data indicates that a proximal region of hMLH1 promoter from −258 to +19 contains _cis_-elements important for driving gene expression. When plasmids pGL3-107, pGL3-175, pGL3-258, pGL3-575, pGL3-734 and pGL3-1045 were used to transfect another colorectal cancer cell line HCT8 which also showed normal hMLH1 expression as HT29 did, a similar luciferase activity pattern was obtained.
Figure 2
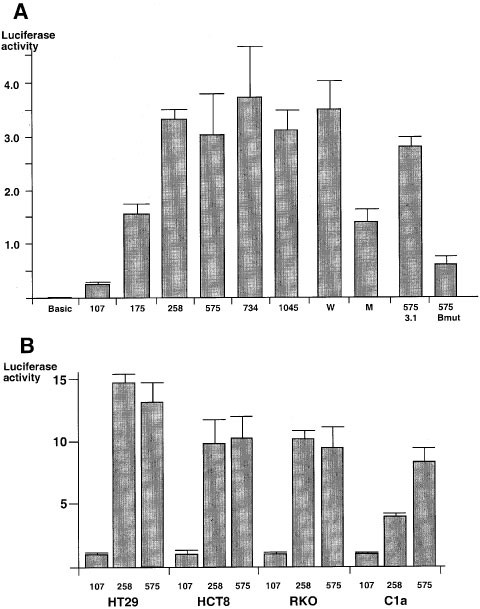
Luciferase activities of the transfectants transfected with plasmid constructs containing different lengths of hMLH1 promoter. (a) HT29 cells were transfected with plasmids pGL3-Basic, pGL3-107, pGL3-175, pGL3-258, pGL3-575, pGL3-734, pGL3-1045, pGL3-W and pGL3-M using transfection reagent Tfx-50. HT29 cells were also co-transfected with pGL3-575 and pcDNA3.1(+) or with pGL3-575 and pcCBF-Bmut. Forty-eight hours after the transfection, luciferase activities were measured by Dual-Luciferase Reporter Assay System. Each column represents the relative luciferase activity from cells transfected with each plasmid, using the luciferase activity from pGL3-Promoter as 1. The luciferase activities were averaged from three different assays, and the bars indicate the standard deviations. (b) Cell lines HT29, HCT8, RKO and C1a were transfected with pGL3-107, pGL3-258 and pGL3-575 as described in a. Each column represents the relative luciferase activity using that from pGL3-107 transfectant from each cell line as 1. The luciferase activities were averaged from three different assays, and the bars indicate the standard deviations
To test the hypothesis that loss of hMLH1 expression is due to the dysfunction of transcription machinery in cells which lack hMLH1 expression, we also transfected two other colorectal cancer cell lines RKO and C1a in which hMLH1 gene is not expressed. To our surprise, high levels of luciferase activities similar to those found in HT29 and HCT8 transfectants were observed in both transfected RKO and C1a cells. In HT29, HCT8, RKO and C1a cells, the luciferase activities in pGL3-258 transfectants were 14.6, 9.8, 10.8 and 3.4 times higher than those in the pGL3-107 transfectants, respectively. The luciferase activities in pGL3-575 transfectants were 14.0, 11.0, 10.1 and 8.1 times higher than those in pGL3-107 transfectants (Figure 2b). This observation indicates that even in RKO and C1a cells that lack hMLH1 expression, the transcription machinery responsible for hMLH1 expression is functionally intact. This also rules out the hypothesis that the lack of hMLH1 expression is due to dysfunctional transcription machinery in non-expressing cell lines.
DNA from the transfected cells was isolated, treated with NaHSO3, and subjected to methylation analysis by COBRA method (Xiong and Laird, 1997). The NaHSO3-treated DNA was amplified by PCR with a primer set, which included an upstream primer with hMLH1 promoter sequence and a downstream primer with sequence from the plasmid so that only the hMLH1 promoter sequence in the plasmid, but not the endogenous hMLH1 promoter was amplified. The transfected hMLH1 promoter sequence (exogenous) showed no methylation not only in HT29 and HCT8 transfectants, but also in C1a and RKO transfectants with methylated endogenous hMLH1 promoter. This observation explains the fact that the luciferase activities in the RKO and C1a transfectants are as high as those in HT29 and HCT8 transfectants, because the transfected promoters (exogenous) are not methylated. We have previously shown that in all tested cell lines, regardless of the hMLH1 expression levels, the transcription machinery is intact. Therefore, in RKO and C1a cells, the functional transcription factors and the unmethylated hMLH1 promoter are capable of driving the expression of the report gene effectively.
To further investigate the role of methylation in regulating gene expression, the plasmids pGL3-258 and pGL3-575 were in vitro methylated by _Hha_I and _Hpa_II methylases (New England Biolabs, Beverly, MA, USA), and transfected into RKO cells. The plasmids were treated with _Hha_I and _Hpa_II methylases according to the procedures recommended by the manufacturers. The completeness of methylation by _Hha_I and _Hpa_II methylases was checked by measuring the extent of protection from digestion of the restriction enzymes _Hha_I and _Hpa_II, respectively. After RKO cells were transfected with the methylated plasmids, the luciferase activities in these transfectants were significantly reduced compared to those found in the cells transfected with the unmethylated pGL3-258 and pGL3-575 (Figure 1b,c). Since the luciferase gene in the plasmids can also be methylated by methylases, we need to distinguish the inhibition of luciferase by the methylation of hMLH1 promoter from that by the methylation of the luciferase. Thus, we used plasmid pGL3-107 as a control, which carries a short proximal promoter sequence without _Hha_I or _Hpa_II sites. The reason why we used pGL3-107 rather than pGL3-Basic as a control is that the former showed more reliable luciferase activities than the latter, because the former contains a short promoter sequence (Figures 1 and 2a). Due to the fact that the methylation at CpG sites in luciferase coding region can inhibit the basal level of gene expression, the treatment with _Hha_I and _Hpa_II methylases reduced the luciferase activities in the methylated pGL3-107 transfectants to 33.8±6.8 and 23.5±4.1% of those of the unmethylated pGL3-107 transfectants, respectively (Figure 1b,c). Since _Hha_I methylase can methylate CpG site in GCGC sequence, after _Hha_I methylase treatment, 2 and 4 CpG sites in the promoter of pGL3-258 and pGL3-575 were methylated, respectively (Figure 1b). The luciferase activities from the methylated pGL3-258 and pGL3-575 were 12.9±0.6 and 11.5±3.7% of those from the unmethylated pGL3-258 and pGL3-575. These retained activities were obviously lower than that from the methylated pGL3-107 (33.8±6.8%). This comparison mentioned above indicates that methylation in the region between −258 and +19 was more effective than the region between −575 and –258 in suppressing the promoter activity by methylation, since methylation in region between −258 and +19 reduced the luciferase from 33.8±6.8 to 12.9±0.6% in _Hha_I methylase treated pGL3-258 transfectant, while two more methylation sites in region between −575 and −258 did not reduce the luciferase activities further (comparing 11.5±3.7 with 12.9±0.6%). _Hpa_II methylase can methylate CpG site in CCGG sequence. Since four CCGG sequences are present only in the region from −575 to −258, four CpG sites in hMLH1 promoter in pGL3-575 were methylated, while no CpG site in pGL3-258 was methylated due to the lack of CCGG sequence (Figure 1c). The retained luciferase activities of the _Hpa_II methylase treated pGL3-258 and pGL3-575 transfectants were 22.1±4.1 and 20.2±3.5% respectively, similar with that of the methylated pGL3-107 transfectants (23.5±4.1%). This observation also proves that methylation in the region between −575 and −258 was not as important as the region between −248 to +19 in inhibiting the expression by methylation.
This conclusion is consistent with our previous observation that methylation in only a small proximal region of hMLH1 (region C, between −248 and −178) invariably correlated with the absence of gene expression, while methylation in upstream regions (regions A and B, between −711 and -266) did not necessarily correlate with the gene silencing (Figure 1a, Deng et al., 1999). We also observed in the previous study that after treating non-hMLH1 expressing cells with a demethylation agent 5-aza-2′-deoxycytidine to induce hMLH1 expression, the extent of demethylation was higher in this proximal region than the more upstream region (Deng et al., 1999). Thus, we concluded that this proximal region plays a major role in regulating hMLH1 expression by methylation.
DNA fragments which cover different regions of hMLH1 promoter from −575 to +19 were used as probe in electrophoretic mobility shift assay (EMSA, Mathey-Prevot et al., 1990) using HeLa cell nuclear proteins (Promega). A major shifted band was detected when a fragment that covered −266 to −227 of the hMLH1 promoter was used. Since this shifted band is the most significant one we detected in the whole region from −575 to +19, and this region is also within the proximal region we have previously identified, we decided to concentrate our EMSA studies in this region. In this region we found a short sequence containing CCAAT box, a motif which a transcription factor CBF (including CBF-A, CBF-B and CBF-C subunits) binds to (Sinha et al., 1996; Currie, 1997; Mantovani, 1998). To study the significance of the CCAAT box, we annealed a pair of complementary oligonucleotides (5′-AACGAACCAATAGGAAGAG-3′ and 5′-TCCGCTCTTCCTATTGGTT-3′) to make a double-stranded probe with 23 base pairs (bp) in length which contained CCAAT box. When this 32P-labeled probe was mixed with the nuclear proteins from HeLa cells, a shifted band was detected (Figure 3a, lane 2, indicated by arrow). The band was competed off by different amounts of unlabeled competitor (Figure 3a, lanes 3–5). Nuclear proteins were also prepared from RKO, C1a, HCT8 and HT29 cells as described previously (Andrews and Faller, 1991). When the nuclear proteins from RKO, C1a, HCT8 and HT29 cells were used in EMSA with the same probe as mentioned above, the same shifted bands were observed (data not shown). This also proves that in all different cell lines, regardless of the hMLH1 expression levels, the transcription factors are functional in binding.
Figure 3

EMSA using nuclear extracts from HeLa cells. (a) Two pmol of a pair of complementary oligonucleotides (5′-AACGAACCAATAGGAAGAG-3′ and 5′-TCCGCTCTTCCTATTGGTT-3′) were heated to 60°C, annealed by cooling to room temperature slowly, and labeled with 32P at 3′ end by repair synthesis with Klenow fragment of DNA Polymerase I (Gibco-BRL, Gaithersburg, MD, USA). In lane 1, 0.05 pmol (>104 c.p.m.) of the probes were incubated in 10 μl containing 10 mM Tris-HC1, pH 7.5, 50 mM NaC1, 5 mM MgC12, 0.5 mM DTT, 10% glycerol, 0.05% NP-40, 0.05 mg/ml poly(dI-dC) at room temperature for 20 min, and separated on a 4% polyacrylamide gel. In lane 2, the probes were incubated in the same solution as in lane 1, except 5 μg of nuclear proteins from HeLa cells were added to the solution. In lanes 3, 4 and 5, unlabeled probes (150, 50 and 15 ng respectively) were added to compete the binding with the labeled probes. Arrow indicates the main band shifted by the binding of nuclear proteins. (b) The wild type probe with CCAAT box was obtained from a pair of complementary oligonucleotides described in a. The mutant probe with TTGGC replacement was obtained from a pair of complementary oligonucleotides: 5′-AACGAATTGGCAGGAAGAG-3′ and 5′-TCCGCTCTTCCTGCCAATT-3′. Wild type probe (lanes 1–5) and mutant probe (lanes 6–10) were incubated with nuclear proteins from HeLa cells (lanes 2–5 and lanes 7–10) as described in a. Antibodies against CBF-A (lanes 3 and 8), CBF-B (lanes 4 and 9) and Sp1 (lanes 5 and 10) were added after the incubation. The mixtures were incubated again at room temperature for 30 min, and separated on a 4% polyacrylamide gel
Antibodies against CBF-A, CBF-B and Sp1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used in super shift assay (Figure 3b). When antibodies against CBF-A and CBF-B were used, a significant shift was observed (lanes 3 and 4) as compared with the sample without antibody (lane 2), while antibody against Sp1 did not change the mobility of the band (lane 5). This indicates that CBF is the transcription factor that binds to CCAAT box. When a mutated probe containing a 5 bp replacement in CCAAT box was used in the super shift assay (Figure 3b, lanes 6-10), no band was detected, indicating CCAAT is the specific sequence for the binding of CBF-A and CBF-B.
From the EMSA and super shift assays, we identified a CCAAT box in the proximal region of hMLH1 promoter, which binds to a well-defined transcription factor CBF (NF-Y). The specificity of the binding was shown by the competition in EMSA (Figure 3a, lanes 3–5). CCAAT is the core sequence for the binding shown by the loss of binding to the mutant probe (Figure 3b, lanes 7–10). In order to study the biological function of the CCAAT box, we constructed a plasmid containing luciferase gene driven by hMLH1 promoter (from −272 to +19) with a mutant CCAAT box replaced by TTGGC sequence, named pGL3-M. HT29 cells were transfected separately with pGL3-M and its wild type counterpart, pGL3-W. The luciferase activity of pGL3-M transfectant was 2.5 times lower than that of pGL3-W transfectant (Figure 2a). This observation indicates that CCAAT box and transcription factor CBF play a role in up-regulating hMLH1 gene expression.
CBF has been proved to regulate the transcription in a variety of genes, such as ferritin (Marziali et al., 1997), myeloperoxidase (Orita et al., 1997), ABO (Kominato et al., 1997), and α2 collagen, E2F1 (Hu and Maity, 2000). The function of CBF and its DNA binding site was shown by transient transfection of reporter containing the wild type and mutant CCAAT boxes, and by EMSA in which CBF bound to either wild type or mutant CCAAT sequences. The biological function of CBF was recently further proved in vivo by analysing the inhibition of reporter activities after co-transfection of a dominant negative mutant CBF-B subunit (Marziali et al., 1997; Hu and Maity, 2000).
We have shown that CBF and its binding to CCAAT box in hMLH1 promoter play a role in regulating gene expression by transfection assay and EMSA. To further prove their function in vivo, we constructed a plasmid, containing a mutant CBF-B subunit. To do this, a plasmid, pFLAG-Bmut, carrying mutant CBF-B at 304th and 311th codons (Hu and Maity, 2000) was amplified by PCR with two primers flanking the coding sequence of CBF-B. The PCR product was then digested with restriction enzymes _Eco_RI and _Xho_I. Since the PCR primers contain _Eco_RI and _Xho_I sites respectively, after digestion with these two enzymes the PCR product could be inserted into an expression vector, pcDNA3.1(+), containing cytomegalovirus enhancer-promoter. The newly constructed plasmid, pcCBF-Bmut, and the plasmid pGL3-575 were used to transfect HT29 cells. The luciferase activity of the pcCBF-Bmut transfectant was 4.4 times lower than that of the pcDNA3.1(+) transfectant (Figure 2a). Since the plasmid pcCBF-Bmut expressed mutant CBF-B subunit, and this DNA binding mutant acted as a dominant repressor of the complex formed by the endogenous wild type CBF and CCAAT box, the transcription of luciferase was thus inhibited (Mantovani et al., 1994). This showed that CBF plays an important role in the activation of hMLH1 gene.
The probe containing CCAAT box used in the previously described EMSA was methylated at two CpG sites by _Sss_I methylase (New England Biolabs, Beverly, MA, USA) following the procedure recommended by the manufacturers. One of the two CpG sites is located 2 bp upstream of CCAAT box, and the other CpG site is 8 bp downstream of CCAAT box. The methylated probe was incubated with the nuclear proteins from HeLa cells (Figure 4). In order to exclude the possibility that the treatment of the probe results in a change in the non-specific binding capacity, we used a mock-treated oligo probe as a control. This control probe was treated with the same methylation procedure except that S-adenosylmethionine was omitted. The binding of the nuclear proteins to the methylated oligo probe was significantly reduced compared with the binding to the mock-treated probe. A 35 bp fragment containing the same CCAAT box was obtained from hMLH1 promoter sequence digested with restriction enzymes _Sac_I and _Sau_3A, and also used as a probe in this comparison. The band from the methylated fragment was also much weaker than that from the mock-treated fragment (Figure 4). This observation shows that the methylation at CpG sites close to CCAAT box interferes with the binding of transcription factor to CCAAT box. Thus, we conclude that in hMLH1 non-expressing cells, the silencing of the transcription is mediated by the inhibition of binding of CBF to CCAAT box, but not by the defective transcription factor.
Figure 4

Comparison of the in vitro methylated and the mock-treated probes in binding capacity by EMSA. The labeling of the oligo probe was described in Figure 3. The 32P-labeled probe was methylated by _Sss_I methylase according to the procedure described by the manufacturers. _Sac_I/_Sau_3A fragment was obtained by digestion of the plasmid pGL3-575 with these two restriction enzymes. Its labeling and methylation were performed as oligo probe. Half of the labeled oligo probe and _Sac_I/_Sau_3A probe were in vitro methylated, the other half oligo and _Sac_I/_Sau_3A probes were treated with the same procedure except the S-adenosylmethionine was omitted. These mock-treated oligo probe and _Sau_I/_Sau_3A fragment were used as the control to compare the nuclear proteins' binding with those to the methylated oligo and _Sac_I/_Sau_3A probes. The main shifted bands are indicated by the arrow
In this study, we have shown by functional assay that a proximal region (−258 to +10) of hMLH1 promoter was capable of driving the transcription, and the promoter activity was significantly inhibited by methylation, especially when the CpG sites in the proximal region were methylated. We observed that in all colorectal cancer cell lines we tested, the luciferase activities were high, regardless of the hMLH1 expression levels. Similarly, in EMSA the nuclear proteins isolated from all colorectal cancer cell lines, including the non-expressing cell lines, bound to CCAAT box efficiently. These observations indicate that the mechanism of the loss of hMLH1 expression in these non-expressing cells is not due to the defect of the transcription machinery. Thus our results suggest that the silencing of transcription is due to the methylation of specific CpG sites which inhibits the binding of transcription factors to the _cis_-elements. This hypothesis was proved by EMSA in our study.
Recently, there is increasing evidence showing that DNA methylation is responsible for gene silencing (Jones and Laird, 1999). There are at least two major hypotheses to explain this mechanism. First, the suppression is mediated by the binding between the methylated DNA and a family of methyl-CpG binding proteins, leading to the changing of the chromatin structure. Second, the silencing is due to the inhibition of the binding of sequence-specific transcription factors to their binding sites containing CpG (Tate and Bird, 1993). Since not many transcription factor binding sites contain CpG, the second mechanism has been thought not to be frequently involved. However, our observation in the present study showed that a CpG site does not need to be part of a transcription factor binding site to affect the binding. Methylation at a CpG site close to CCAAT box can effectively interfere with the binding of CBF, leading to the suppression of hMLH1 expression. The inhibitory effect of the methylation at CpG sites close to CCAAT box may be mediated by induction of the changes in the DNA double helix configuration, or by interfering with the binding of co-factors, that may inhibit CBF binding indirectly.
In our previous study, we have shown by NaHSO3-sequencing that in hMLH1 promoter, methylation at only certain CpG sites (not every CpG site) is critical (Deng et al., 1999). Similar observations have been made by other researchers with the MGMT and FHIT genes (Qian and Brent, 1997; Tanaka et al., 1998). In this study, the reduction of luciferase activities driven by the promoter with mutated CCAAT box further proves that methylation in a region with CCAAT box plays an important role in hMLH1 regulation.
To understand the mechanisms involved in hMLH1 silencing is very important for the investigation of carcinogenesis of colorectal cancer with MSI, and for the translational application to early diagnosis and prevention of colorectal cancer. We have shown for the first time that methylation of a CpG site close to CCAAT box inhibits the binding of CBF to CCAAT box, resulting in the inhibition of the transcription of hMLH1. However, additional factors, such as transcription factors, activators, chromatin remodelers should also be considered in the regulation of hMLH1 gene in cancer cells.