The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum (original) (raw)
Two basic approaches have been taken to genome sequencing. The statistical approach (‘shotgun sequencing’) relies on the determination of a highly redundant set of random genomic DNA sequences, which are assembled in the computer, the gaps remaining to be closed by other methods5. This approach rapidly yields 90–98% of the genomic information, but requires an extensive robotic infrastructure. The directed approach relies on a complete mapping of the genome, followed by the sequencing of large overlapping genomic fragments by shotgun sequencing or primer walking6. This approach reduces the infrastructure requirements but is slowed down by the need for a genetic map.
One of our aims was to establish a strategy for sequencing microbial genomes in reasonable time without extensive infrastructure. This strategy (‘shotgun primer walking’) combines features of the statistical and directed methods. After construction of several phagemid libraries and one cosmid library, inserts from randomly chosen clones were sequenced from the ends by primer walking (see Supplementary Information). Sequencing was stopped in regions that were redundant with already determined sequences. In total, 400 phagemids, covering 850 kilobase-pairs (kbp) (54%), and 469 cosmids, covering 1,533 kbp (98%), were partially or fully sequenced. With an average insert length of 40 kbp, the clones of the cosmid library statistically covered the genome 12 times. Because cosmids are susceptible to recombination events, the reverse strand of cosmid DNA was always sequenced with DNA templates from other cosmid clones covering the same region, or from polymerase chain reaction (PCR) fragments generated from genomic DNA. As the project advanced, fragments were assembled into progressively larger contigs, until 4 gaps of 33 kbp in total were left. These were closed by four PCR fragments, which were sequenced by primer walking. By this method, we completed the 1.56 Mbp genome of T. acidophilum with 7,855 sequencing reactions. For comparison, shotgun sequencing of the 1.86 Mbp Thermotoga maritima genome, which has the same G+C content, required over 30,000 sequencing reactions.
During the annotation process, we detected ten open reading frames (ORFs) with putative frameshifts. The corresponding genomic regions were amplified by PCR and resequenced with the primers obtained during primer walking. Four of the frameshifts were confirmed, and six had to be corrected. All errors occurred in phagemids, which, unlike cosmids, were sequenced on both strands but without using template DNA from other sources. This suggests that in genome projects operating with low sequence coverage the two strands should be sequenced with template DNA from different sources. It also shows, however, that high-quality data can be obtained with only twofold sequence coverage.
Beyond the advantages discussed here, shotgun primer walking generates an extensive primer set, covering the whole genome. This represents a powerful tool, which can be used to determine rapidly from PCR fragments differences in related DNA sequences, for example between strains of pathogenic bacteria or between individuals in human populations.
The genome of T. acidophilum DSM 1728, one of the smallest among free-living organisms, consists of a single circular chromosome of 1,564,905 bp (Table 1). We did not detect any plasmids, either by biochemical methods or through DNA sequencing. A single plasmid of 15.2 kbp has previously been reported in other isolates of T. acidophilum7. The origin of replication was estimated using the cumulative skew of the orientation of ORFs as well as different nucleotide skews8. The centre of a 477-bp intergenic region in this area was chosen as nucleotide 1.
Table 1 Genome features
Thermoplasma contains one copy of each of the ribosomal RNAs, which are dispersed in the genome9 and separated by at least 52 kbp. This is different from all other known archaea, where at least two of the three genes are contiguous on the chromosome.
We identified 1,509 ORFs in the genome (see Supplementary Information), of which over one-third have homologues in all three domains of life (see Supplementary Information). After manual curation, 823 ORFs (55%) were considered similar to functionally annotated proteins, 446 (29%) resembled hypothetical ORFs in other organisms, and 240 (16%) were not recognizably similar to other sequences in current databases (‘singletons’). In total, we could assign 685 ORFs (46%) to categories of the MIPS Functional Catalogue (see Supplementary Information ). The only category that deviates significantly from other archaea is transport facilitation, which occupies a larger proportion of the genome than in any other archaeon, but is not exceptional when compared with some bacteria (Thermotoga, Deinococcus). By conservative search methods, we could match 620 domains of 537 ORFs (35.6%) to proteins of known structure (Table 1; Supplementary Information ). The distribution of structural classes resembles that of other prokaryotic genomes, but shows a striking overrepresentation of three-layer (αβα) sandwich folds (43% of all matches), which primarily encompass enzymes.
About one-third of the ORFs (484; 32%) occurred in 139 conserved gene clusters, as judged relative to a set of 7 bacterial and 6 archaeal genomes (see Methods). Twenty-two of these clusters occurred only in Thermoplasma and bacteria and are presumably due to lateral transfer. The clusters occur in ‘hot spots’ on the chromosome (Fig. 1), suggesting that entire gene regions may have been acquired in discrete genetic events. The largest detected clusters encode ribosomal proteins (Ta1249–Ta1271), NADH dehydrogenase subunits (Ta0959–Ta0970), precorrin biosynthesis proteins (Ta0653–Ta0660) and flagellar proteins (Ta0553–Ta0560).
Figure 1: Chromosomal distribution of Thermoplasma ORFs most similar to Sulfolobus (top panel), and of ORFs found in conserved gene clusters (bottom panel).
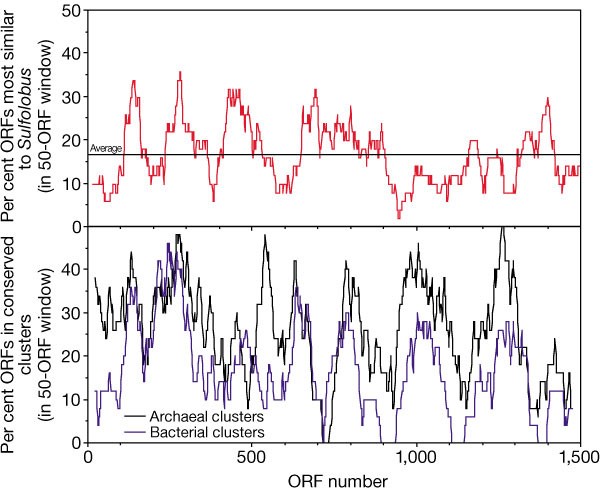
ORFs were considered to occur in a conserved cluster if their chromosomal vicinity was also observed in at least 1 of 13 complete reference genomes (6 archaeal, 7 bacterial; see Methods). The approximate locations of proteins most similar between Thermoplasma and Sulfolobus mentioned in the text are indicated in the top panel.
In the thermoacidophiles Thermoplasma and Sulfolobus, glucose degradation proceeds by a non-phosphorylated variant of the Entner–Doudoroff pathway10 (see Supplementary Information), in which the first step is catalysed by glucose dehydrogenase. The acetyl-CoA produced in this pathway enters the oxidative tricarboxylic acid (TCA) cycle. A complete set of TCA enzymes is recognizable in Thermoplasma, many of which have been studied experimentally. In the glycolysis/gluconeogenesis pathway (Embden–Meyerhof–Parnas; EMP), homologues are recognizable for most enzymes, but not for phosphofructokinase and fructose bisphosphate aldolase, so the presence of a working EMP pathway11 cannot be confirmed.
Thermoplasma is microaerophilic and contains several respiratory chain proteins, such as electron transfer flavoproteins (Ta0429, Ta0329 and Ta0212) and cytochrome b homologues (Ta1222, Ta1228 and Ta1003). Two of the latter are found in gene clusters with Rieske iron–sulphur proteins (Ta1223 and Ta1229). In addition, we identified two homologues of a cytochrome bd quinol oxidase12 (Ta1484 and Ta0992).
Thermoplasma is also able to gain energy anaerobically by sulphur respiration2. Unexpectedly, no homologues of the genes that mediate sulphur respiration in Archaeoglobus fulgidus13 were found. Instead, Thermoplasma contains homologues of AsrA (Ta0046) and AsrB (Ta0047), which mediate dissimilatory sulphur reduction in Salmonella typhimurium14. A sulphide-quinone reductase homologue (Ta1129) may also be involved in sulphur respiration.
Most studied archaea are motile and previous work suggested that they use a chemotaxis signal transduction pathway resembling that of bacteria15. However, Thermoplasma is flagellated and motile2, yet lacks recognizable chemotaxis proteins. This situation is also found in other archaea (Methanococcus jannaschii, Aeropyrum pernix) and bacteria (Aquifex aeolicus), suggesting the presence of an alternative, unrelated signal transduction pathway. Another puzzle concerning motility is the lack of a cell wall, which anchors the flagella in prokaryotes. It is unclear what structure could function as the stator for flagellar rotation in Thermoplasma.
Two chaperones of Thermoplasma have been studied experimentally, the thermosome3 and the protein ‘unfoldase’ VAT16,17. Additional chaperones recognizable in the genome ( Table 2) include three Hsp20-like proteins (Ta0437, Ta0471 and Ta0864) and a complete Hsp60 system, consisting of two thermosome subunits (α, Ta0980; and β, Ta1276) and two prefoldin/Gim subunits (α, Ta1076; and β, Ta1137). Unlike many archaea, Thermoplasma also contains an Hsp70 system (DnaK, Ta1087; DnaJ, Ta1088; and GrpE, Ta1086), whose genes are clustered on the chromosome in contrast to the endogenous Hsp60 genes.
Table 2 The chaperone complement of T. acidophilum
Thermoplasma contains several ‘unfoldases’ (AAA+ proteins)17, which are frequently associated with proteolytic degradation18. These are the Cdc48 homologues VAT (Ta0840) and VAT-2 (Ta1175), an archaeal-type Lon protease (Ta1081) and a Lon-related ATPase lacking the protease domain (Ta0098). Surprisingly, Thermoplasma is missing the proteasomal AAA ATPase (PAN; ref. 19), which has hitherto been found in all archaea. It is conceivable that Ta0098, VAT-2 or VAT, which has protein unfolding activity17, can substitute for this function.
Other chaperones include two protein disulphide isomerases (Ta0125 and Ta0866), a peptidyl-prolyl isomerase of the FKBP family (Ta1011) and two homologues of cold-shock protein CsaA (Ta1284, Ta1046). Ta1284 is most similar to the carboxy-terminal domain of archaeal methionyl-tRNA synthetases (MetRS); this domain is missing from Thermoplasma MetRS. Overall, a surprising number of chaperones from Thermoplasma appear to have been acquired from bacteria (Table 2).
The pathway for polypeptide degradation in Thermoplasma is well understood20. Unfolded proteins are cleaved into fragments of 6–15 residues by the proteasome (α, Ta1288; and β, Ta0612), whose structure and activity have been extensively studied4. These fragments are further degraded by the tricorn peptidase (Ta1490) to di-, tri- and tetrapeptides, which are hydrolysed to free amino acids by three tricorn-interacting factors F1 (Ta0830), F2 (Ta0301) and F3 (Ta0815). It is, however, unclear how proteins are targeted to this degradation pathway, because ubiquitin, whose existence in T. acidophilum had been proposed on the basis of peptide sequences21, is not present in this genome (or indeed in any other archaeon). It is also unclear how proteins are unfolded before degradation by the proteasome, as Thermoplasma is missing a proteasomal AAA ATPase.
In addition to the 6 proteins involved in this degradation pathway, Thermoplasma contains at least 23 putative proteases ( Table 3, Fig. 2), as well as several α/β hydrolases of unknown specificity and a broad-spectrum deacetylase with potential glutamate carboxypeptidase activity (Ta0934). Few proteases seem to be of bacterial origin; however, nine are most similar to proteins of the phylogenetically distant crenarchaeon Sulfolobus solfataricus (http://niji.imb.nrc.ca/sulfhome/results.html). These proteins are tricorn, its interacting factors and five extracellular, multidomain acid proteases (Ta1403, Ta0167, Ta0376, Ta0741 and Ta0976), three of which are membrane-anchored by C-terminal transmembrane sequences. It is unclear how Thermoplasma retains unanchored secreted proteins without a cell wall, but we note that the two secreted proteases and one of the anchored ones (Ta0167) contain a domain conserved in many archaeal S-layer proteins, which may serve as a scaffold.
Table 3 The protease complement of T. acidophilum
Figure 2: Overview of the main proteolytic pathways in Thermoplasma.
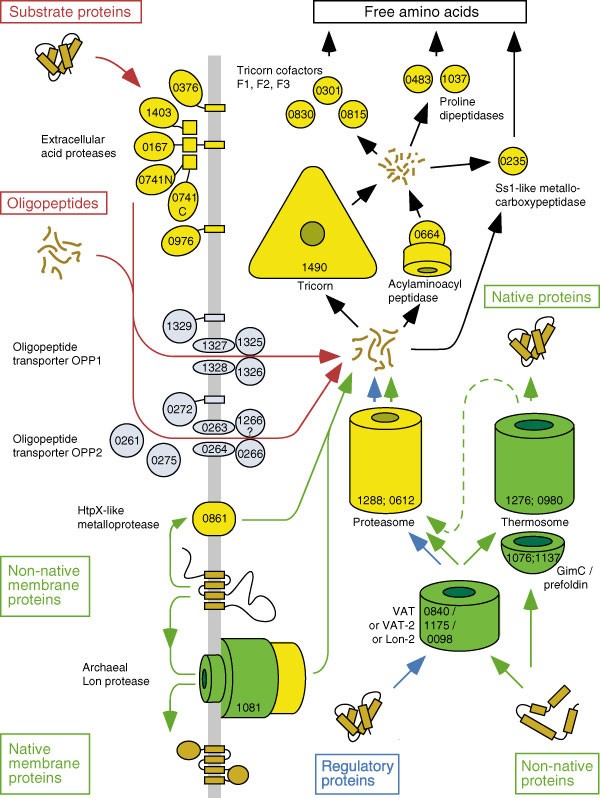
Red arrows indicate the metabolic pathway, green arrows the ‘quality control’ pathway for damaged proteins and blue arrows the regulatory pathway. Chaperones are shown in green, proteases in yellow and transporters in blue. The numbers indicate the ORF code; that is, 1490 corresponds to Ta1490.
Overall, ten Thermoplasma proteases are probably extracellular. These include two membrane-anchored signal peptidase I homologues (Ta0151 and Ta0378), which are closely related to their eukaryotic homologues and may yield useful model systems for the more elaborate eukaryotic complex, a membrane-anchored serine peptidase of the S49 family (Ta1083), a probable _O_-sialoglycoprotein endopeptidase (Ta0324) and a metalloprotease of the M6 family (Ta0728). The two latter proteins are probably secreted, on the basis of experimental findings from other organisms. Like their homologues22, however, they lack recognizable signal sequences. In total, 11 Thermoplasma proteases are membrane-associated, including the archaeal Lon protein, which is anchored to the membrane from the cytoplasmic side by a helical hairpin, a structure present in all archaea but missing in bacterial proteins. As the membrane-bound protease FtsH, which is essential in bacteria, is absent from archaea, its function may be assumed by the archaeal Lon protease.
As discussed, Thermoplasma surprisingly lacks ubiquitin, a proteasomal ATPase and an archaeal-type sulphur respiration system. We searched systematically for further differences to other archaeal genomes using clusters of orthologous groups (COGs; http://www.ncbi.nlm.nih.gov/COG). The first four published archaeal genomes share a ‘stable core’ of 543 COGs; thirty-six of these appear to be missing in Thermoplasma (http://www.biochem.mpg.de/baumeister/genome). The most notable are DNA topoisomerase VI and eukaryal-like histones. Topoisomerase VI (ref. 23) is a divergent type II topoisomerase present only in archaea. In its place, Thermoplasma contains a bacterial-type DNA gyrase (GyrA, Ta1054; and GyrB, Ta1055).
Thermoplasma also contains proteins not present in any other archaeal genome (68, after elimination of singletons; http://www.biochem.mpg.de/baumeister/genome). Most noteworthy was Hta (Ta0093), the first archaeal DNA-binding protein to be investigated24, which is closely related to bacterial proteins and appears to substitute functionally for the missing histones. Other proteins include an egghead homologue (Ta0213), a membrane-anchored protein kinase (Ta0488) and a Ras-like GTPase (Ta1192).
In the past, a _Thermoplasma_-like organism has been debated as a possible ancestor of the eukaryotic cytoplasm. The genome sequence, however, shows that Thermoplasma is a typical archaeon with a fairly large protein complement of bacterial origin. We could not identify any of the proteins peculiar to eukaryotes, such as subunits of the nuclear pore complex, the exocyst or the cytoskeleton. However, Thermoplasma has been reported to contain a cytoskeleton25, and we identified in genome searches a cytosolic coiled-coil protein (Ta1488), which is conserved in all archaea and seems analogous to intermediate filament proteins.
During the comparison to other archaea we noted that 252 Thermoplasma ORFs (17%) were most similar to proteins of S. solfataricus, a higher number than for any other organism in our reference set—even though this genome sequence is still incomplete. Of these, 60 appear to function in transport and are frequently clustered (for example, Ta1325–Ta1329, Ta0261–Ta0275 and Ta0126–Ta0146). Many others are involved in metabolism (including energy metabolism) or in proteolysis, or are secreted or membrane-bound ORFs of unknown function. With a few notable exceptions, such as DNA polymerase (Ta0450) or Rad50 (Ta0157), hardly any are involved in information processes (replication, transcription and translation). Although it has been observed previously that in prokaryotes core information processing generally tracks organism phylogeny, whereas metabolism is strongly affected by lateral transfer, it is highly unusual to see such a large number of genes transferred between two phylogenetically distant organisms. We propose that the adaptation to an extreme environment shared by few other organisms has led to substantial genetic exchange. This may have proceeded by only a few large genetic events, resulting in the skewed distribution of Sulfolobus -like genes in the Thermoplasma genome (Fig. 1).
Thermoplasma inhabits a hot and highly acidic environment, sometimes as low as pH 0.5, in which few organisms are viable. It has adapted to scavenging nutrients from the decomposition of organisms killed by the extreme acidity and requires yeast, bacterial or meat extract when grown in culture. There is an absolute growth requirement for basic oligopeptides26 and analysis of the genome shows the presence of a complete degradation chain for exogenous polypeptides, starting with a set of large extracellular proteases, over two oligopeptide transport systems, to a cytosolic degradation machine consisting of tricorn and its cofactors (Fig. 2). All proteins of this degradation chain are most similar to cognate proteins of Sulfolobus, as are many other proteins involved in transport and metabolism. Thus, it would seem that two classes of genes can be distinguished in Thermoplasma: One is primarily composed of constitutive, ‘housekeeping’ genes, which generally reflect the phylogenetic origin of the organism. The other mostly contains ‘life-style’ genes, including but not limited to metabolism, which are tailored to a specific environment and are shared between phylogenetically distant organisms within one ecological niche.