Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling (original) (raw)
Abstract
Studies of the actin-based motility of the intracellular pathogens Listeria monocytogenes and Shigella flexneri have provided important insight into the events occurring at the leading edges of motile cells1,2,3,4,5. Like the bacteria Listeria and Shigella, vaccinia virus, a relative of the causative agent of smallpox, uses actin-based motility to spread between cells6. In contrast to Listeria or Shigella, the actin-based motility of vaccinia is dependent on an unknown phosphotyrosine protein, but the underlying mechanism remains obscure7. Here we show that phosphorylation of tyrosine 112 in the viral protein A36R by Src-family kinases is essential for the actin-based motility of vaccinia. Tyrosine phosphorylation of A36R results in a direct interaction with the adaptor protein Nck8 and the recruitment of the Ena/VASP family member N-WASP9 to the site of actin assembly. We also show that Nck and N-WASP are essential for the actin-based motility of vaccinia virus. We suggest that vaccinia virus spreads by mimicking the signalling pathways that are normally involved in actin polymerization at the plasma membrane.
Similar content being viewed by others
Main
We began our investigation into the mechanism of actin-based motility of vaccinia virus by identifying the phosphotyrosine protein observed at the site of actin assembly7. Western-blot analysis reveals that three prominent proteins, pTyr200, pTyr80/85 and pTyr50, consistently become tyrosine phosphorylated during vaccinia infection (Fig. 1). Western-blot analysis and immunoprecipitation experiments identified pTyr80/85 as the actin-binding protein cortactin10 (data not shown), which is known to be enriched in the cell cortex and becomes phosphorylated by c-Src when Shigella enters the cell11. To determine whether tyrosine phosphorylation of pTyr200, pTyr80/85 and pTyr50 was dependent on actin tail formation, we examined the phosphorylation pattern of cells infected with the recombinant viral strains ΔA34R, ΔA36R and ΔF13L, which do not induce actin tails at 8 hours post-infection12,13. We found that pTyr50 was absent in cells infected with a recombinant virus lacking A36R (Fig. 1). Western-blot analysis of two-dimensional gels and immunoprecipitation experiments showed that pTyr50 and A36R are the same protein (data not shown). The vaccinia A36R protein is a type Ib integral membrane protein with a cytoplasmic domain of about 200 amino-acid residues13 that plays an essential but undefined role in the actin-based motility of vaccinia12,14.
Figure 1: Vaccinia A36R protein is tyrosine phosphorylated.
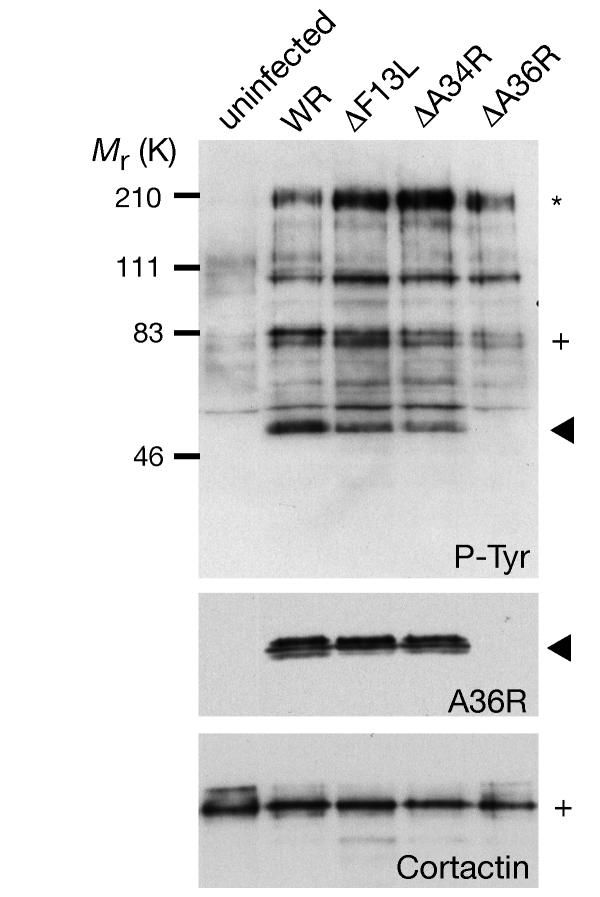
Top, western-blot analysis of phosphotyrosine proteins during vaccinia infection reveals that three proteins, pTyr200 (asterisk), pTyr80/85 (cross) and pTyr50 (arrowhead) consistently become tyrosine phosphorylated, although pTyr50 (arrowhead) is absent in ΔA36R infections. Bottom, the same blot probed with A36R and cortactin antibodies. Virus strains are indicated.
To study the role of A36R phosphorylation in vaccinia actin-based motility, we tested whether ectopic expression of the wild-type A36R protein in ΔA36R infected cells would rescue actin tail formation. Ectopic expression of A36R resulted in both tyrosine phosphorylation of the protein and efficient rescue of actin tail formation (111 ± 36%) (Fig. 2a, c). Expression of point mutants of the six individual tyrosine residues in the A36R cytoplasmic domain showed that only a change of tyrosine 112 to phenylalanine (A36R mutant Y112F) resulted in a dramatic reduction of actin tails (Fig. 2b). However, western-blot analysis revealed that mutation of either tyrosine 112 or tyrosine 132 reduced A36R phosphorylation, indicating that both of these residues are phosphorylated (Fig. 2c). As A36R–Y112F still rescues actin tail formation, albeit at low levels, we produced the double point mutant, YdF, in which both residues 112 and 132 were changed to phenylalanine. Although expression of A36R–YdF was similar to the other point mutants, we found no evidence for either tyrosine phosphorylation of the protein or rescue of actin tail formation (Fig. 2b, c). Lack of rescue was not due to mistargeting, as A36R–YdF localized to the intracellular enveloped form of vaccinia virus, which is responsible for actin tail formation6 (data not shown). In parallel with studies using A36R point mutants, we examined the ability of several A36R deletion constructs to rescue actin tail formation (Fig. 2d). The only requirement for actin tail formation was the presence of tyrosine 112, which represents a good Src-family kinase phosphorylation site15. Vaccinia infection also induces phosphorylation of cortactin, a well known Src substrate10, so we tested whether this family of kinases phosphorylates A36R. We found that the Src-family kinase-inhibitor PP1 (ref. 16) reduced tyrosine phosphorylation of both A36R and cortactin, as well as reducing actin tail formation (Fig. 3a; and data not shown). However, PP1 did not affect viral assembly or A36R localization (data not shown). In addition, expression of c-Src kinase ‘dead open’ mutants 527 Kin- and KP Kin- reduced tyrosine phosphorylation of A36R and actin tail formation (Fig. 3a, b; and data not shown). When expressed at low levels, 527 Kin- and KP Kin- were observed at the site of vaccinia actin tail formation, which is consistent with their dominant-negative activity when overexpressed (Fig. 3c).
Figure 2: Tyrosine phosphorylation of A36R is essential for vaccinia actin tail formation.
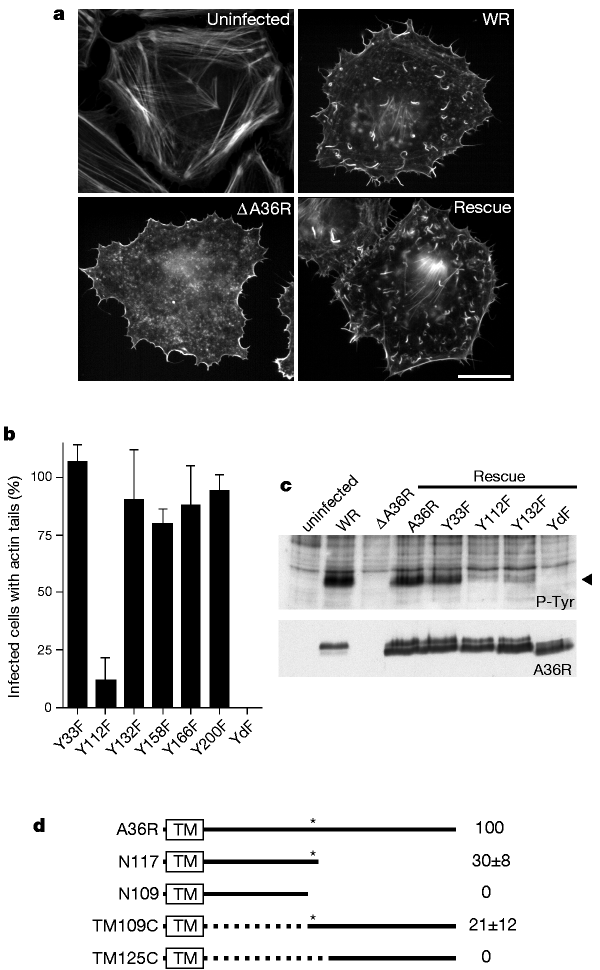
a, Immunofluorescence analysis reveals that ectopic expression of A36R in the ΔA36R background rescues actin tail formation. Scalebar, 20 µm. b, Quantification of actin tail formation by all six A36R tyrosine-to-phenylalanine mutants reveals that only Y112 is required for efficient actin tail formation. However, only the A36R–YdF double mutant (Y112F and Y132F combined) eliminates all actin tail formation. c, Top, western-blot analysis of ectopically expressed A36R point mutants reveals that ΔA36R-infected cells expressing Y112F or Y132F have a weaker pTyr50 signal than cells infected with WR, or with ΔA36R expressing wild-type A36R or Y33F. Y158F, Y166F and Y200F mutants are phosphorylated to a similar extent as A36R or Y33F (data not shown). No pTyr50 signal is detectable in ΔA36R-infected cells expressing the YdF double mutant. Bottom, the same blot reprobed with anti-A36R antibody. YdF migrates slightly faster than the other A36R proteins, consistent with an absence of tyrosine phosphorylation. d, Diagram of A36R and the deletion mutants N109, N117, TM109C and TM125C (see Methods for nomenclature). The box at the N terminus shows the predicted transmembrane domain (TM). Asterisk indicates Y112; dotted line indicates the region of A36R that is deleted in TM109C and TM125C; numbers on the right show the rescue efficiencies of actin tail formation.
Figure 3: Src-family kinases mediate actin tail formation of vaccinia virus.
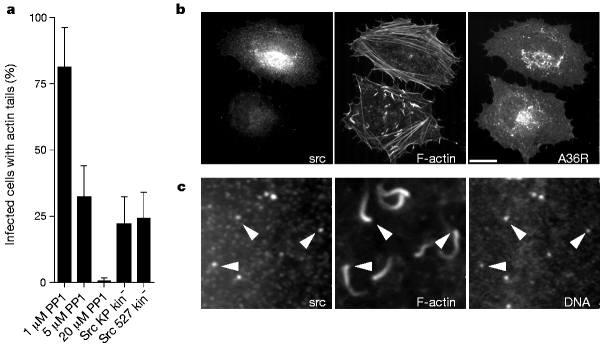
a, The Src-family kinase inhibitor PP1 inhibits actin tail formation in a dose-dependent manner. Overexpression of dead open c-Src mutants also reduces the efficiency of actin tail formation. b, Immunofluorescence analysis of the effects of overexpressing dead open c-Src mutants on vaccinia actin tail formation. Two infected cells, one transfected (top) and one untransfected (bottom), are shown. Scale bar, 20 µm. c, Closer examination of infected cells expressing low levels of dead open c-Src reveals that the protein is recruited to viral particles that have induced actin tails (arrowheads).
Our observations that vaccinia actin tail formation is dependent on Src-family kinases and tyrosine phosphorylation is reminiscent of signal-transduction pathways involved in the control of actin polymerization at the plasma membrane. This analogy is strengthened by the localization of the adaptor protein Nck8 at the site of vaccinia actin tail formation (Fig. 4a). Nck recruitment was dependent on both A36R and its phosphorylation, as it was absent in ΔA36R infections, with or without ectopic expression of A36R–YdF (data not shown). In vitro binding assays using peptides corresponding to residues 105–116 of A36R demonstrate that recruitment of Nck to the virus occurs through the direct interaction with A36R and is dependent on phosphorylation of tyrosine 112 (Fig. 4b). Furthermore, overexpression of the Nck SH2 domain resulted in a dramatic reduction of actin tail formation (Fig. 4c). The involvement of Nck raises the question whether its downstream partner WASP8,17,18 is also required for vaccinia actin tail formation. We found that N-WASP9, a member of the Ena/VASP family, is recruited to the virus (Fig. 4a). N-WASP was also efficiently recruited from cell extracts by the Nck–A36R-phosphorylated peptide complex (Fig. 4b). As observed with the Nck SH2 domain, overexpression of a dominant-negative construct, N-WASP–ΔWA19, inhibits actin tail formation (Fig. 4c). The requirement of N-WASP, which is known to stimulate the actin-nucleating activity of the Arp2/3 complex20,21, seems to provide the final link to achieve vaccinia actin tail formation. Nck and N-WASP, together with the Arp2/3 complex, are also observed on clathrin-coated vesicles that can nucleate little actin tails7 (Fig. 4), indicating that this complex of proteins plays an important role in initiating actin polymerization in the cell.
Figure 4: Nck and N-WASP are essential for vaccinia actin tail formation.
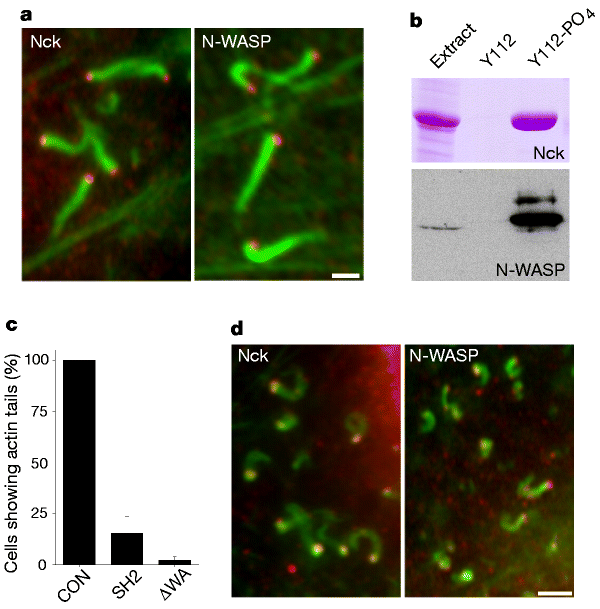
a, Immunofluorescence images showing that Nck and N-WASP (red) are recruited to vaccinia virus particles that have induced actin tails (green). Scale bar, 2 µm. b, Top, Coomassie-stained gel showing that the peptide A36R105-116 containing phosphorylated tyrosine 112 interacts directly with Nck, as it retains the protein from soluble Escherichia coli extract. Y112 and Y112-PO4 represent the unphosphorylated and phosphorylated versions of the A36R105-116 peptide, respectively. Bottom, western blot showing that the Nck-A36R105-116 complex recruits N-WASP from extracts prepared from infected cells. The resin sample represents five times more material that the extract sample, demonstrating there is no recruitment of N-WASP by the unphosphorylated A36R105-116 peptide. c, Overexpression of the Nck-SH2 domain (SH2) or an N-WASP construct lacking the WA domain (ΔWA) inhibits actin tail formation in WR-infected cells. CON, control cells infected with WR but not transfected. d, Immunofluorescence images showing that Nck and N-WASP (red) are recruited to clathrin-coated vesicles that can nucleate actin tails (green) in uninfected cells. Scale bar, 2 µm.
Vaccinia virus therefore achieves actin-based motility by mimicking the receptor tyrosine kinase signalling pathways that control actin polymerization at the plasma membrane. The sequence conservation between vaccinia A36R and its homologue A39R in variola virus, the causative agent of smallpox, indicates that variola virus also used actin-based motility to spread between cells. This Src-family kinase-dependent signalling pathway may also account for other cellular effects induced by vaccinia infection, including induction of cell motility, loss of contact inhibition and changes in cell adhesion, all of which are reminiscent of a transformed phenotype22,23. We suggest that vaccinia virus provides an excellent model system in which to study Src-family kinase and the signalling pathways that affect actin polymerization, adhesion and cell motility.
Methods
Western-blot and immunofluorescence analysis
Western reserve and recombinant vaccinia strains that lack the genes A34R, A36R or F13L are referred to as WR, ΔA34R, ΔA36R and ΔF13L12. HeLa cells were infected and processed for western-blot or immunofluorescence analysis, and images were acquired as described7,13. Intracellular enveloped virious were labelled with A33R or A36R antibodies13, and tyrosine-phosphorylated proteins were detected with the 4G10 monoclonal antibody (Upstate Biotechnology). Src was detected by using anti-Src 327 mouse monoclonal antibody24. Antibodies against cortactin and NcK were obtained from Upstate Biotechnology, Transduction Laboratories and Santa Cruz Biotechnology. Rhodamine- and Alexa 488-phallacidin and PP1 were obtained from Molecular Probes and Alexis Corporation, respectively.
Construction of pEL A36R expression vectors
A36R expression was placed under the control of the synthetic vaccinia virus early/late promoter (E/L)25 by polymerase chain reaction (PCR) using the primers PELA36RFor 5′-CCGCCGCTCGAGAAAAATTGAAATTTTATTTTTTTTTTTTGGAATATAAATAAGCTCGAGATCTACCATGATGCTGGTACCTCTTATC-3′ and A36RH3Rev 5′-GGCCAAGCTTATCACACCAATGATACGACCGATGATTCTAT-3′. The resulting PCR product was cloned into the _Xho_I–_Hin_dIII sites of _Not_I-deficient pBluescript SKII to generate the pELA36R construct. Tyrosine-to-phenylalanine point mutations of A36R were engineered by ‘round the world’ PCR26 using pELA36R as a template. The pELA36R–YdF construct was engineered in the same way using A36R–Y112F as the template. The deletion mutants N109 and N117 were generated by introducing a stop codon after residue 109 and 117, respectively, and are named according to the remaining sequence, that is, amino-terminal to their final carboxy-terminal residue (Fig. 2d). We took advantage of a unique SnaB1 site, which immediately follows the transmembrane (TM) domain of A36R (residues 1–32) to generate internal deletions by PCR. Internal deletions TM109C and TM125C are defined by the presence of the transmembrane domain followed by the first residue of the cytoplasmic domain of A36R (109 or 125) extending to the C terminus (Fig. 2d).
pEL dominant-negative expression constructs
_Eco_RI–_Bam_HI inserts containing the c-Src mutants 527 Kin- and KP Kin- were cloned into the _Eco_RI–_Bgl_II sites of the pELA36R vector backbone. The 527 and KP of the K295M catalytically dead (Kin-) c-Src represent mutations of Y527F and K249G/P250E24, respectively. These proteins represent ‘dead open’ conformations of c-Src which are able to bind cellular targets but are catalytically inactive. EGFP was amplified by PCR from the pEGFP-N1 expression vector (Clontech) and cloned into the _Bgl_II–_Xho_I sites of pEL immediately downstream of the viral promoter. The DNA coding residues 1–397 of rat N-WASP9 were then amplified by PCR and cloned into the _Not_I–_Eco_RI sites of the pELGFP vector to generate pELGFP N-WASP–ΔWA, which lacks the WH2 and acidic domains of the protein19. The DNA corresponding to the SH2 domain, residues 275–373, of human Nck1 and the full-length protein were amplified by PCR from a HeLa cDNA library and cloned in an identical fashion to N-WASP–ΔWA into pELGFP. The fidelity of all pEL constructs was confirmed by sequencing.
Expression and quantification of actin tail rescue or inhibition
pEL constructs were transfected into HeLa cells 4 hours post-infection with either WR or ΔA36R viral strains and processed for immunofluorescence or western-blot analysis 4 hours later. Similarly, PP1 was added to cells either before infection or up to 4 hours post-infection. Rescue of actin tail formation or inhibition efficiencies were determined from over 200 infected cells in 4 independent experiments based on criteria described previously7. All data were normalized to WR or A36R rescue experiments where appropriate, and error bars indicate standard deviation of the mean.
Protein expression and in vitro binding
Human Nck1 was cloned from pELGFP into the vector pMW172, expressed in BL21(DE3), and the soluble fraction prepared as described27. The tyrosine-phosphorylated and unphosphorylated versions of the peptide CGGAPSTEHIYDSVAGST, corresponding to residues 105–116 of A36R, were coupled by the N-terminal cysteine residue to Sulfolink resin (Pierce). The BL21(DE3) soluble fraction containing Nck1 was incubated with peptide resin in the presence of 1 mM vanadate at room temperature for 30 min and washed 5 times with phosphate-buffered saline containing 100 mM NaCl, 0.1% Tween and 1 mM vanadate. The washed resin was then incubated with cell extracts prepared as described13. Samples were removed for SDS–PAGE and western-blot analysis to assess Nck1 and N-WASP binding.
References
- Dramsi,S. & Cossart,P. Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 14, 137–166 (1998).
CAS PubMed Google Scholar - Finlay,B. B. & Cossart,P. Exploitation of mammalian host cell functions by bacterial pathogens. Science 276, 718–725 (1997).
CAS PubMed Google Scholar - Welch,M. D., Iwamatsu,A. & Mitchison,T. J. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385, 265–269 (1997).
ADS CAS PubMed Google Scholar - Welch,M. D., Rosenblatt,J., Skole,J., Portnoy,D. A. & Mitchison,T. J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 281, 105–108 (1998).
ADS CAS PubMed Google Scholar - Mullins,D. R., Heuser,J. A. & Pollard,T. D. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl Acad. Sci. USA 95, 6181–6186 (1998).
ADS CAS PubMed PubMed Central Google Scholar - Cudmore,S., Cossart,P., Griffiths,G. & Way,M. Actin-based motility of vaccinia virus. Nature 378, 636–638 (1995).
ADS CAS PubMed Google Scholar - Frischknecht,F. et al. Tyrosine phosphorylation is required for actin based motility of vaccinia but not Listeria or Shigella. Curr. Biol. 9, 89–92 (1999).
CAS PubMed Google Scholar - McCarty,J. H. The Nck SH2/SH3 adaptor protein: a regulator of multiple intracellular signal transduction events. BioEssays 20, 913–921 (1998).
CAS PubMed Google Scholar - Miki,H., Minura,K. & Takenawa,T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 15, 5326–5335 (1996).
PubMed PubMed Central Google Scholar - Wu,H. & Parsons,J. T. Cortactin, an 80/85-kilodalton pp60sc substrate, is a filamentous actin-binding protein enriched in the cell cortex. J. Cell Biol. 120, 1417–1426 (1993).
CAS PubMed Google Scholar - Dehio,C., Prevost,M. C. & Sansonetti,P. J. Invasion of epithelial cells by Shigella flexneri induces tyrosine phosphorylation of cortactin by a pp60c-src-mediated signalling pathway. EMBO J. 14, 2471–2482 (1995).
CAS PubMed PubMed Central Google Scholar - Sanderson,C. M., Frischknecht,F., Way,M., Hollinshead,M. & Smith,G. L. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 79, 1415–1425 (1998).
CAS PubMed Google Scholar - Röttger,S., Frischknecht,F., Reckmann,I., Smith,G. L. & Way,M. Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J. Virol. 73, 2863–2875 (1999).
PubMed PubMed Central Google Scholar - Wolffe,E. J., Weisberg,A. S. & Moss,B. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 25, 20–26 (1998).
Google Scholar - Songyang,Z. & Cantley,L. C. Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends Biochem. Sci. 20, 470–475 (1995).
CAS PubMed Google Scholar - Hanke,J. H. et al. Discovery of a novel, potent, and src family selective tyrosine kinase inhibitor. J. Biol. Chem. 271, 695–701 (1996).
CAS PubMed Google Scholar - Rivero-Lezcano,O. M. & Marcilla,A., Sameshima,J. H. & Robbins,K. C. Wiskott-Aldrich syndrome protein physically associates with Nck through Src3 homology domains. Mol. Cell. Biol 15, 5725–5731 (1995).
CAS PubMed PubMed Central Google Scholar - Snapper,S. B. & Rosen,F. S. The Wiskott-Aldrich syndrome protein (WASP): roles in signalling and cytoskeletal organization. Annu. Rev. Immunol. 17, 905–929 (1999).
CAS PubMed Google Scholar - Machesky,L. M. & Insall,R. H. Scar1 and the related Wiskott-Aldrich syndrome protein WASP regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8, 1347–1356 (1998).
CAS PubMed Google Scholar - Machesky,L. M. et al. Scar, a WASp-related protein, activates dendritic nucleation of actin filaments by the Arp2/3 complex. Proc. Natl Acad. Sci. USA 96, 3739–3744 (1999).
ADS CAS PubMed PubMed Central Google Scholar - Rohatgi,R. et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221–231 (1999).
CAS PubMed Google Scholar - Sanderson,C. M., Way,M. & Smith,G. L. Virus-induced cell motility. J. Virol. 72, 1235–1243 (1998).
CAS PubMed PubMed Central Google Scholar - Sanderson,C. M. & Smith,G. L. Vaccinia virus induces calcium-independent cell-matrix adhesion during the motile phase of infection. J. Virol. 72, 9924–9933 (1998).
CAS PubMed PubMed Central Google Scholar - Gonfloni,S. et al. The role of the linker between the SH2 domain and catalytic domain in the regulation and function of Src. EMBO J. 16, 7261–7271 (1997).
CAS PubMed PubMed Central Google Scholar - Chakrabarti,S., Sisler,J. R. & Moss,B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 23, 1094–1097 (1997).
CAS PubMed Google Scholar - Hemsley,A., Arnheim,N., Toney,M. D., Cortopassi,G. & Galas,D. J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 17, 6545–6551 (1989).
CAS PubMed PubMed Central Google Scholar - Way,M., Pope,B. & Weeds,A. G. Evidence for functional homology in the F-actin binding domain of gelsolin and alpha-actinin: implications for the requirements of severing and capping. J. Cell Biol. 119, 835–842 (1992).
CAS PubMed Google Scholar
Acknowledgements
We are grateful to H. Miki for N-WASP cDNA and N-WASP antibody. We thank G. Smith and R. Blasco for providing recombinant vaccinia strains; J. White, C. Blaumüller, A. Desai and A. Ploubidou for reading the manuscript; T. Harder for encouraging us to try PP1; and S. Guth for the ‘round the world’ PCR method. S.G. was supported by a fellowship from the Boncompagni-Ludovisi Foundation.
Author information
Authors and Affiliations
- Cell and Developmental Biology Programmes, European Molecular Biology Laboratory, Meyerhofstrasse 1, Heidelberg, 69117, Germany
Friedrich Frischknecht, Violaine Moreau, Sabine Röttger, Stefania Gonfloni, Inge Reckmann, Giulio Superti-Furga & Michael Way
Authors
- Friedrich Frischknecht
You can also search for this author inPubMed Google Scholar - Violaine Moreau
You can also search for this author inPubMed Google Scholar - Sabine Röttger
You can also search for this author inPubMed Google Scholar - Stefania Gonfloni
You can also search for this author inPubMed Google Scholar - Inge Reckmann
You can also search for this author inPubMed Google Scholar - Giulio Superti-Furga
You can also search for this author inPubMed Google Scholar - Michael Way
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMichael Way.
Rights and permissions
About this article
Cite this article
Frischknecht, F., Moreau, V., Röttger, S. et al. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling.Nature 401, 926–929 (1999). https://doi.org/10.1038/44860
- Received: 23 June 1999
- Accepted: 19 August 1999
- Issue Date: 28 October 1999
- DOI: https://doi.org/10.1038/44860