Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas (original) (raw)
- Mini Review
- Open access
- Published: 17 May 2011
Minireview
British Journal of Cancer volume 104, pages 1805–1809 (2011)Cite this article
- 2644 Accesses
- 75 Citations
- 3 Altmetric
- Metrics details
Subjects
This article has been updated
Abstract
Local recurrence of glioblastomas is a major cause of patient mortality after definitive treatment. This review discusses the roles of the chemokine stromal cell-derived factor-1 and its receptor CXC chemokine receptor 4 (CXCR4) in affecting the sensitivity of glioblastomas to irradiation. Blocking these molecules prevents or delays tumour recurrence after irradiation by inhibiting the recruitment of CD11b+ monocytes/macrophages that participate in revascularising the tumour. We review the literature pertaining to the mechanism by which revascularisation occurs following tumour irradiation using experimental models. Areas of interest and debate in the literature include the process by which endothelial cells die after irradiation and the identity/origin of the cells that reconstitute the tumour blood vessels after injury. Understanding the processes that mediate tumour revascularisation will guide the improvement of clinical strategies for preventing recurrence of glioblastoma after irradiation.
Similar content being viewed by others
Main
Despite advances in techniques for administering radiotherapy, local recurrence of glioblastomas (GBMs) typically leads to patient mortality. In this review, we focus on the chemokine stromal cell-derived factor-1/CXC chemokine ligand 12 (SDF-1/CXCL12) and its receptor CXCR4 in affecting the sensitivity of GBM to irradiation. We begin with an overview of the discovery and characterisation of these molecules, followed by an examination of their role in tumour metastasis and tissue injury. Finally, we discuss the data and controversies surrounding their role in revascularisation and tumour recurrence. Our understanding of the role of SDF-1 and CXCR4 in tumour recurrence post irradiation is largely gained from pre-clinical studies using mouse xenografts of human tumours. The pre-clinical data will be the focus of this review. Clinical studies in this area are limited, but serve as an exciting area for future investigation.
The CXC chemokine family has multiple roles in leukocyte migration, immune response, angiogenesis, and tumour metastasis. CXC chemokines have four highly conserved cystine amino-acid residues, with a non-conserved amino acid separating the first two cystine residues. A second structural domain determines their function. The CXC chemokine family contains both angiogenesis-promoting and angiogenesis-inhibiting molecules. Stromal cell-derived factor-1 is regarded as an angiogenesis-promoting chemokine.
Stromal cell-derived factor-1 is a small pro-inflammatory chemoattractant cytokine that was first cloned by Tashiro et al, 1993, as a protein captured by its specific amino-terminal signal sequences. It was later identified functionally as a molecule involved in B-cell lymphopoiesis using an expression cloning strategy. Human CXCR4 was initially identified as a receptor for SDF-1 by screening chemokine receptor orphan genes for their ability to induce intracellular Ca2+ in response to human SDF-1. The mouse CXCR4 receptor was subsequently found by cloning candidate chemokine receptors and comparing the amino acid sequence with the human cDNA (Nagasawa et al, 1998). Recently, it has been shown that SDF-1 binds to another receptor, CXCR7 (Burns et al, 2006). This finding has opened up new possibilities for understanding the biology of SDF-1 and modulating SDF-1-mediated pathways.
Mice with genetic deletion of SDF-1 or CXCR4 are embryonic lethal and have defects in embryogenesis, haematopoiesis, cardiac ventricular septum formation, migration of neural precursors in the cerebellum, and blood vessel formation in the gastrointestinal tract (Nagasawa et al, 1998; Tachibana et al, 1998). The similar phenotypes of these mice suggest that CXCR4 may be the critical receptor through which SDF-1 acts during development. In the adult mouse, SDF-1 expression is found in lymphoid (bone marrow, thymus, and spleen) and non-lymphoid organs (brain, heart, lung, liver, kidney, stomach, and intestine). In the blood, CXCR4 expression has been described in peripheral blood monocytes, neutrophils, lymphocytes, and haematopoietic progenitors (Nagasawa et al, 1998), and has been shown to be involved in cell migration. Stromal cell-derived factor-1 has also been shown to induce migration of brain microglia and astrocytes (Tanabe et al, 1997). In the adult, SDF-1 functions as a chemoattractant for haematopoietic cells (Aiuti et al, 1997). In addition to its role in chemotaxis, the SDF-1/CXCR4 pathway has been reported to be a survival factor for human glioma cells through its upregulation of the PI3K/AKT pathway (Zhou et al, 2002).
CXC chemokine receptor 4 is a seven-transmembrane G-protein-coupled receptor that acts as a receptor for SDF-1 and is also a co-receptor for HIV entry into target cells. A discussion of CXCR4's role in HIV is beyond the scope of this review. We will instead briefly review what is known about signal transduction downstream of CXCR4 on binding SDF-1. Binding of SDF-1 to CXCR4 leads to phosphorylation of ERK2 and protein kinase B, coupled with activation of PI3-kinase and formation of PIP3 (Tilton et al, 2000). Stromal cell-derived factor-1-mediated signalling through CXCR4 has also been shown to mediate phosphorylation of focal adhesion proteins such as RAFTK, paxillin, and Crk, and transcriptional activation through p44/42 MAP kinase (Erk 1 and 2), and activation of NF-_κ_B (Ganju et al, 1998). As our current understanding of intracellular signalling pathways downstream of CXCR4 is largely through in vitro studies, it is not well understood to what extent these multiple signal transduction pathways are shared among the different cell types or between normal vs malignant cells.
The SDF-1/CXCR4 pathway in tumour metastasis and tissue injury
The interaction between the CXCR4 receptor and its ligand, SDF-1_α_, has been shown to mediate tumour metastasis (Muller et al, 2001; Sun et al, 2005). Overexpression of CXCR4 has been detected in a variety of human malignancies, including breast, pancreatic, prostate cancer, and GBM. Muller et al (2001) demonstrated that CXCR4 is expressed in primary breast cancer cells and that SDF-1 was highly expressed in the most common sites of metastasis, including the lymph nodes, lungs, liver, and bone. When the CXCR4/SDF-1 interaction was blocked with a neutralising anti-CXCR4 antibody using an in vivo xenotransplant model, metastatic load was significantly reduced (Muller et al, 2001). These data support the role of the SDF-1/CXCR4 pathway in the formation of metastasis for some tumours.
The SDF-1/CXCR4 pathway is also involved in tissue repair (Kollet et al, 2003; Ratajczak et al, 2003). Kollet et al (2003) showed that CCl4-mediated liver injury led to an increase in the recruitment of human CD34+ progenitor cells by SDF-1 to the injured liver in NOD/SCID mice, suggesting that SDF-1 may direct haematopoietic progenitor cells to sites of tissue injury. However, whether these cells in the liver represent functional haematopoietic stem/progenitor cells (capable of reconstituting lethally irradiated hosts) was not examined. Chemotaxis of CXCR4-expressing murine muscle satellite cells towards SDF-1 has also been demonstrated, suggesting that migration of tissue-specific stem cells may be regulated by SDF-1 (Ratajczak et al, 2003).
The role of SDF-1/CXCR4 in restoring functional vasculature in irradiated GBMs
Radiotherapy has a crucial role in the treatment of GBMs, but despite the very high radiation doses that can be delivered with new techniques such as high-dose boost stereotactic radiosurgery, the tumours invariably recur, leading to mortality in 75% of patients by 2 years after initial diagnosis. As most of the recurrences occur within the radiation field (Minniti et al, 2010; McDonald et al, 2010), any method of improving local control of the primary tumour by radiotherapy would improve the curability of patients with GBM. We have recently proposed that the cure rates for this malignancy might be improved by targeting the SDF-1/CXCR4 pathway, thereby preventing reconstitution of the tumour vasculature following irradiation.
In theory, reestablishing the tumour vasculature after radiation could be mediated either by sprouting angiogenesis (from adjacent blood vessels) or by circulating cells – including endothelial cells (ECs) or endothelial progenitor cells (EPCs), a process known as vasculogenesis (see the section ‘Restoration of tumour blood vessels after irradiation’). In pre-clinical models, several investigators have reported that radiation inhibits local angiogenesis, the generation of new vessels from surrounding vessels (Udagawa et al, 2007; Imaizumi et al, 2010). However, in these studies it is difficult to determine whether cells, locally or distantly, mediate tumour revascularisation after irradiation. To interrogate whether new vasculature formation relies on a different mechanism in the presence vs the absence of radiation, we examined the effect of radiation on the development of tumour vasculature in the absence of matrix metalloproteinase-9 (MMP-9), a key proangiogenic molecule in circulating CD11b+ cells. We demonstrated that tumours cannot grow in an irradiated site (given 20 Gy) of an MMP-9 knockout (KO) mouse but can grow in a non-irradiated MMP-9 KO mouse. Tumour growth is restored following irradiation if the bone marrow in the MMP-9 KO mouse is replaced with wild-type bone marrow (Ahn and Brown, 2008). Thus, MMP-9 from cells in the bone marrow transplant could restore tumour vasculature (determined by CD31 immunostaining and injection of Hoechst dye) and support tumour growth at a pre-irradiated site. This illustrated that revascularisation after irradiation required extracellular matrix modelling of MMP-9 by cells in the bone marrow, although tumour growth without irradiation did not, suggesting that they may depend on different pathways for recruiting new vasculature. We demonstrated through depletion experiments and immunostaining that CD11b+ cells mediate this effect. It is important to note that our finding that radiation prevents local angiogenesis is not the same as the proposal of Fuks and Kolesnick that radiation produces a rapid apoptosis of tumour ECs and vascular shutdown (Garcia-Barros et al, 2003). We and others who have measured tumour ECs after irradiation do not see this rapid apoptosis (Kioi et al, 2010; Kozin et al, 2010). Rather, we observe that ECs disappear slowly after irradiation over several days, consistent with a mitotically linked death.
CD11b+ monocytes/macrophages are important mediators of tumour revascuzlarisation and regrowth after irradiation. Neutralising the antibodies against CD11b+ monocytes/macrophages given after irradiation markedly enhances the antitumour effect of radiation in human tumour xenografted mice (Ahn et al, 2010). We observe that these monocytes also express CXCR4 on their surface. Inhibition of the interaction of SDF-1 and CXCR4 using the CXCR4 inhibitor, AMD3100, or antibodies against CXCR4 prevented the radiation-induced increase of CD11b+ monocytes/macrophages and inhibited revascularisation in intracranial human GBM xenografts. Moreover, these inhibitors abrogated or delayed the recurrence of the tumours following irradiation (Kioi et al, 2010). Potentiation of the anticancer drug BCNU by AMD3100 has also been shown with the intracranially implanted U87 human glioblastoma, though the mechanism for this may have involved abrogation of the survival function of the SDF-1/CXCR4 pathway in the tumour cells (Redjal et al, 2006) rather than prevention of revascularisation after irradiation.
We hypothesise that elevated levels of tumour SDF-1 caused by increased tumour hypoxia (resulting from gradual loss of ECs) following irradiation lead to accumulation of CXCR4-expressing monocytes/macrophages in the irradiated tumour. This is also suggested by data in the literature showing that the recruitment and retention of proangiogenic hematopoietic cells to sites of ischaemic tissue damage or to tumours is mediated by the interaction of the SDF-1 with CXCR4 (Ceradini et al, 2004; Aghi et al, 2006; Jin et al, 2006). Stromal cell-derived factor-1 functions as a hypoxia-inducible gene through the action of the transcription factor hypoxia inducible factor-1 (HIF-1). We have shown that irradiated tumours gradually lose vasculature after irradiation, thereby becoming increasingly hypoxic and upregulating HIF-1. Consistent with this, the HIF-1 inhibitor NSC 134754 prevents both the radiation-induced tumour accumulation of CD11b+ monocytes/macrophages and prevents tumour recurrence (Kioi et al, 2010). We and others have shown that SDF-1 levels are increased in irradiated tumours (Kioi et al, 2010; Kozin et al, 2010) and in the plasma following local tumour irradiation of intracranial GBM (unpublished data). A model showing the pathway by which CD11b+ monocytes/macrophages accumulate in irradiated tumours is shown in Figure 1. Also shown in Figure 1 are the points in the pathway that have been targeted therapeutically to prevent the regrowth of irradiated tumours in pre-clinical studies.
Figure 1
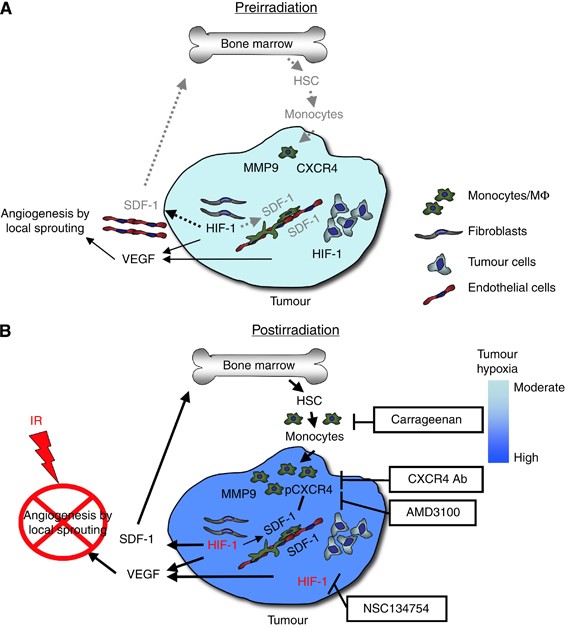
Model of the main contributions of bone marrow-derived cells (BMDCs) and cytokines that promote restoration of tumour vasculature following irradiation. Prior to irradiation, tumour growth is governed largely by local angiogenesis. When local angiogenesis is inhibited by irradiation, growth of the tumour vasculature (essential for recurrence of the tumour) can only occur from circulating cells, of which BMDCs are an essential component. Following irradiation, the tumour becomes more hypoxic and HIF-1 is increased as the tumour attempts to regrow. This induces SDF-1 and promotes the recruitment of CD11b+ monocytes/macrophages and retention of these cells in the tumour. Stromal cell-derived factor-1/CXC chemokine receptor 4 is the key interaction for the influx of BMDCs as AMD3100, an inhibitor of CXCR4/SDF-1, and antibodies against CXCR4 block the recruitment and/or tumour retention of the BMDCs, inhibit restoration of the tumour vasculature, and prevent tumour recurrence. The various inhibitors and the points in the cycle at which they act are shown in boxes. Reproduced from Kioi et al (2010) with permission. (A) Pre-irradiation; (B) post-irradiation.
Restoration of tumour blood vessels after irradiation
Vasculogenesis is a term used in embryology to denote the de novo formation of blood vessels. Its use in the present context would imply that all the cellular components of the tumour vasculature after irradiation come from circulating cells, not from residual vascular cells in the tumour that survive radiation, nor from surrounding angiogenic vessels. We hypothesise that ECs do not regrow from surviving ECs in the radiation field at the doses used in our studies (15–20 Gy) or at TCD50 doses (doses that control 50% of the tumours) typical for transplanted tumours (40–100 Gy), and particularly in SCID mice in which all the stromal cells are highly radiosensitive (Budach et al, 1993).
Experimental data show the importance of bone marrow-derived CD11b+ monocytes/macrophages for the restoration of tumour blood vessels after irradiation. Unlike CD11b+ monocytes/macrophages in non-irradiated tumours, these cells express the angiopoietin-2 receptor Tie-2 as well as MMP-9 and the F4/80 macrophage marker, and are therefore classified as Tie-2-expressing macrophages (TEMs). CD11b+ cells from an irradiated tumour share many of the same properties as ECs and have been shown to interact with newly forming tumour blood vessels (De Palma et al, 2005). Whether or not there exists EPCs in the BM that directly give rise to tumour endothelium has been a source of controversy and confusion in the literature. To address whether cells in the bone marrow directly give rise to the components of blood vessels, we carefully examined the ECs in irradiated tumours in mice with Lac-Z or GFP-expressing bone marrow. From such studies, we and others concluded that the ECs in the tumour do not arise from cells in the bone marrow (Ahn and Brown, 2008; Kioi et al, 2010; Kozin et al, 2010). To our knowledge, the so-called bone marrow-derived ‘endothelial progenitor cells’ have not been shown directly to form tumour vasculature. For instance, Purhonen et al (2008) have shown using a parabiotic mouse system (two mice joined so as to have a common blood supply) that VEGFR-2+ bone marrow cells did not incorporate into the tumour endothelium. Other investigators have shown using either orthotopic aortic allografting (Hillebrands et al, 2002) or a parabiotic mouse system combined with reverse bone marrow transplantation (Aicher et al, 2007) that non-bone marrow-derived circulating ECs or EPCs from the liver and small intestine home to sites of active angiogenesis. Thus, circulating ECs, which are not derived from cells of the adult bone marrow, may be recruited to the irradiated tumour. We hypothesise therefore that circulating ECs or EPCs migrate to the irradiated tumour, thereby reconstituting the vasculature. Such a possibility would fit the existing data but needs further experimental studies to validate the hypothesis. The stimulus for this colonisation by ECs is likely to be SDF-1 via its receptor CXCR4 (Orimo et al, 2005) and/or its receptor CXCR7, which is highly expressed in tumour-associated blood vessels including those in GBM (Miao et al, 2007; Liu et al, 2010). CXCR7 has also recently been shown to be co-expressed with CXCR4 on cortical interneurons and to be essential for their SDF-1-stimulated migration (Sanchez-Alcaniz et al, 2011). CD11b+ cells in the irradiated tumour may also help recruit ECs into the tumour or help reorganise the tumour extracellular matrix via MMP-9 to mediate revascularisation. Thus, the restoration of the tumour vasculature following radiation doses similar to those used in cancer treatment could formally fit the definition of vasculogenesis (i.e., derived entirely from circulating cells), but further work is required to prove this conclusively. Other groups have explored alternative hypotheses by which tumours establish and/or maintain their vasculature, including endothelial differentiation of GBM stem-like cells (Ricci-Vitiani et al, 2010). However, markers used to define ECs can be expressed by normal stem cells and cancer stem cells, and it does not necessarily indicate that GBM stem cells possess plasticity towards an EC fate. A subset of GBM stem cells may acquire features shared by ECs when grown under EC-promoting conditions in vitro, although this may not reflect their behaviour in vivo. Our lab has been focusing on the mechanism of revascularisation in the post-irradiated GBM tumour, as this can be interrupted to dramatically improve the outcome in pre-clinical studies. We are actively interested in identifying the source of ECs in the post-irradiated GBM tumour.
Clinical data
Our data suggest that the pathway of post-irradiation recruitment of CD11b+ monocytes/macrophages in tumours may be important not only in mice but also in human patients. We have shown by immunohistochemistry that the levels of CD11b+ monocytes/macrophages are higher in GBM recurrences than in the same tumours prior to therapy (Kioi et al, 2010). Whether or not these cells mediate revascularisation of human GBM after irradiation is not yet known. However, understanding the pathway involved in local recurrence of GBM after radiotherapy holds much clinical potential for preventing recurrence of GBM.
Recently, a clinical study of 95 patients with GBM reported recurrence in 79 patients (83%) after a median follow-up period of more than 18 months. Although the majority of recurrences were located in the radiation field, in 17 patients GBM recurred outside the radiation field. As recurrences outside the radiation field were observed after a longer follow-up time, different mechanisms may be responsible for in-field and out-of-field recurrences (Minniti et al, 2010). Blocking tumour revascularisation after irradiation by inhibiting vasculogenesis would be the most relevant method to prevent recurrences of GBM in the radiation field and this strategy would benefit the majority of patients with GBM. However, if out-of-field recurrences became the prominent pattern of recurrence after local tumour irradiation, whole-brain irradiation could be given, as this has been a viable clinical option for GBM in the past. Improved markers or imaging for identifying patients at risk for in-field vs out-of-field recurrence would allow us to select patients who would benefit from targeted radiation vs whole-brain irradiation. Preventing revascularisation of tumours after irradiation would be important for both radiation treatment strategies.
Conclusions
Stromal cell-derived factor-1 is a small pro-inflammatory chemoattractant cytokine that binds to its G-protein-coupled receptor CXCR4. The interaction of SDF-1 with CXCR4 has been shown to play a role in tumour metastasis by CXCR4-expressing tumour cells migrating to normal tissues expressing SDF-1. In tissue remodelling after injury, haematopoietic cells migrate to sites of ischaemic injury, where increased levels of SDF-1 are produced by the hypoxic upregulation of HIF-1. It has recently become apparent that migration and recruitment of circulating proangiogenic monocytes/macrophages can occur in tumours following local irradiation. We have proposed that the increased hypoxia seen in tumours following irradiation recruits CD11b+ monocytes/macrophages and ECs to the tumour, thereby restoring the tumour vasculature. The reliance of the tumour on revascularisation after irradiation suggests a promising therapeutic approach involving inhibition of this pathway. There are potentially a number of ways to achieve this, including inhibition of HIF-1, antibodies against CD11b or against CXCR4, and pharmacological inhibition of the SDF-1/CXCR4 pathway with the drug AMD3100 (plerixafor). These strategies have been successfully used in pre-clinical studies. Of these, the use of plerixafor is currently the most promising for further investigations in human clinical trials as a proof of principle, as this drug is already approved for clinical use. Understanding the mechanism by which tumours revascularise after irradiation offers promising therapeutic strategies for treating GBMs.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
- Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA (2006) Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res 66 (18): 9054–9064
Article CAS Google Scholar - Ahn GO, Brown JM (2008) Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell 13 (3): 193–205
Article CAS Google Scholar - Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM (2010) Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA 107 (18): 8363–8368
Article CAS Google Scholar - Aicher A, Rentsch M, Sasaki K, Ellwart JW, Fandrich F, Siebert R, Cooke JP, Dimmeler S, Heeschen C (2007) Nonbone marrow-derived circulating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res 100 (4): 581–589
Article CAS Google Scholar - Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC (1997) The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J Exp Med 185 (1): 111–120
Article CAS Google Scholar - Budach W, Taghian A, Freeman J, Gioioso D, Suit HD (1993) Impact of stromal sensitivity on radiation response of tumors. J Natl Cancer Inst 85 (12): 988–993
Article CAS Google Scholar - Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203(9): 2201–2213
Article CAS Google Scholar - Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10 (8): 858–864
Article CAS Google Scholar - De Palma M, Venneri MA, Galli R, Sergi LS, Politi LS, Sampaolesi M, Naldini L (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8 (3): 211–226
Article CAS Google Scholar - Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE (1998) The alpha-chemokine, stromal cell-derived factor-1alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem 273 (36): 23169–23175
Article CAS Google Scholar - Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R (2003) Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 300 (5622): 1155–1159
Article CAS Google Scholar - Hillebrands JL, Klatter FA, van Dijk WD, Rozing J (2002) Bone marrow does not contribute substantially to endothelial-cell replacement in transplant arteriosclerosis. Nat Med 8 (3): 194–195
Article Google Scholar - Imaizumi N, Monnier Y, Hegi M, Mirimanoff RO, Ruegg C (2010) Radiotherapy suppresses angiogenesis in mice through TGF-betaRI/ALK5-dependent inhibition of endothelial cell sprouting. PLoS One 5 (6): e11084
Article Google Scholar - Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S (2006) Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 12 (5): 557–567
Article CAS Google Scholar - Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120 (3): 694–705
Article CAS Google Scholar - Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, Dabeva MD, Kahn J, Spiegel A, Dar A, Samira S, Goichberg P, Kalinkovich A, Arenzana-Seisdedos F, Nagler A, Hardan I, Revel M, Shafritz DA, Lapidot T (2003) HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest 112 (2): 160–169
Article CAS Google Scholar - Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG (2010) Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 70 (14): 5679–5685
Article CAS Google Scholar - Liu Y, Carson-Walter EB, Cooper A, Winans BN, Johnson MD, Walter KA (2010) Vascular gene expression patterns are conserved in primary and metastatic brain tumors. J Neurooncol 99 (1): 13–24
Article CAS Google Scholar - McDonald MW, Shu HK, Curran Jr WJ, Crocker IR (2010) Pattern of failure after limited margin radiotherapy and temozolomide for glioblastoma. Int J Radiat Oncol Biol Phys 79: 130–136
Article Google Scholar - Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, Howard MC, Schall TJ (2007) CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA 104 (40): 15735–15740
Article CAS Google Scholar - Minniti G, Amelio D, Amichetti M, Salvati M, Muni R, Bozzao A, Lanzetta G, Scarpino S, Arcella A, Enrici RM (2010) Patterns of failure and comparison of different target volume delineations in patients with glioblastoma treated with conformal radiotherapy plus concomitant and adjuvant temozolomide. Radiother Oncol 97 (3): 377–381
Article CAS Google Scholar - Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410 (6824): 50–56
Article CAS Google Scholar - Nagasawa T, Tachibana K, Kishimoto T (1998) A novel CXC chemokine PBSF/SDF-1 and its receptor CXCR4: their functions in development, hematopoiesis and HIV infection. Semin Immunol 10 (3): 179–185
Article CAS Google Scholar - Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121 (3): 335–348
Article CAS Google Scholar - Purhonen S, Palm J, Rossi D, Kaskenpaa N, Rajantie I, Yla-Herttuala S, Alitalo K, Weissman IL, Salven P (2008) Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci USA 105 (18): 6620–6625
Article CAS Google Scholar - Ratajczak MZ, Majka M, Kucia M, Drukala J, Pietrzkowski Z, Peiper S, Janowska-Wieczorek A (2003) Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells 21 (3): 363–371
Article CAS Google Scholar - Redjal N, Chan JA, Segal RA, Kung AL (2006) CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin Cancer Res 12 (22): 6765–6771
Article CAS Google Scholar - Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468 (7325): 824–828
Article CAS Google Scholar - Sanchez-Alcaniz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, Lopez-Bendito G, Stumm R, Marin O (2011) Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron 69 (1): 77–90
Article CAS Google Scholar - Sun YX, Schneider A, Jung Y, Wang J, Dai J, Cook K, Osman NI, Koh-Paige AJ, Shim H, Pienta KJ, Keller ET, McCauley LK, Taichman RS (2005) Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res 20 (2): 318–329
Article CAS Google Scholar - Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393 (6685): 591–594
Article CAS Google Scholar - Tanabe S, Heesen M, Yoshizawa I, Berman MA, Luo Y, Bleul CC, Springer TA, Okuda K, Gerard N, Dorf ME (1997) Functional expression of the CXC-chemokine receptor-4/fusin on mouse microglial cells and astrocytes. J Immunol 159 (2): 905–911
CAS PubMed Google Scholar - Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T (1993) Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science 261(5121): 600–603
Article CAS Google Scholar - Tilton B, Ho L, Oberlin E, Loetscher P, Baleux F, Clark-Lewis I, Thelen M (2000) Signal transduction by CXC chemokine receptor 4. Stromal cell-derived factor 1 stimulates prolonged protein kinase B and extracellular signal-regulated kinase 2 activation in T lymphocytes. J Exp Med 192 (3): 313–324
Article CAS Google Scholar - Udagawa T, Birsner AE, Wood M, D’Amato RJ (2007) Chronic suppression of angiogenesis following radiation exposure is independent of hematopoietic reconstitution. Cancer Res 67 (5): 2040–2045
Article CAS Google Scholar - Zhou Y, Larsen PH, Hao C, Yong VW (2002) CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem 277 (51): 49481–49487
Article CAS Google Scholar
Author information
Authors and Affiliations
- Department of Radiation Oncology, Division of Radiation and Cancer Biology, Stanford University, 269 Campus Drive West, CCSR Room 1255, Stanford, 94305, CA, USA
D Tseng, D A Vasquez-Medrano & J M Brown
Authors
- D Tseng
You can also search for this author inPubMed Google Scholar - D A Vasquez-Medrano
You can also search for this author inPubMed Google Scholar - J M Brown
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJ M Brown.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tseng, D., Vasquez-Medrano, D. & Brown, J. Targeting SDF-1/CXCR4 to inhibit tumour vasculature for treatment of glioblastomas.Br J Cancer 104, 1805–1809 (2011). https://doi.org/10.1038/bjc.2011.169
- Received: 01 September 2010
- Revised: 12 April 2011
- Accepted: 20 April 2011
- Published: 17 May 2011
- Issue Date: 07 June 2011
- DOI: https://doi.org/10.1038/bjc.2011.169