NEMO/IKKγ regulates an early NF-κB-independent cell-death checkpoint during TNF signaling (original) (raw)
Main
A major quest in TNF signaling has been to understand the molecular mechanism that determines whether the survival or the death pathway is triggered after receptor ligation.1, 2 Studies over the past decade have led to the model that it is the activation of the NF-_κ_B signaling pathway on TNF receptor 1 (TNFR1)3 ligation that acts as a cell-death determinant because NF-_κ_B transcription factors induce the expression of antiapoptotic genes. It is also known that the components of the TNFR1 death pathway are preexisting and do not require de novo protein synthesis. In fact historically, apoptosis is induced in most cell types by treating the cells with a combination of TNF and cycloheximide, a protein synthesis inhibitor. Thus the apoptotic pathway does not require new protein synthesis whereas the NF-_κ_B-dependent survival pathway requires transcription and translation of new proteins, and yet most cells are largely resistant to apoptosis when treated with TNF alone. This paradoxical situation suggests that there must be additional mechanisms that keep the apoptotic pathway in check before NF-_κ_B-mediated induction of antiapoptotic genes.
A central regulatory step in NF-κ_B signaling is the activation of the I_κ_B kinase (IKK) complex. The IKK complex, which consists of the catalytic subunits IKK_α and IKK_β_ and the regulatory subunit NEMO, phosphorylates the inhibitory molecule I_κ_B resulting in its degradation and the translocation of NF-_κ_B transcription factors to the nucleus. In the absence of NEMO, the IKK complex cannot be activated and the canonical NF-_κ_B signaling pathway is blocked.3 We and others have previously shown that NEMO-deficient cells are highly sensitive to TNF-induced cell death.4, 5, 6 Because of the essential role of NEMO in IKK activity, it has been assumed that the sensitivity to TNF-induced apoptosis in NEMO-deficient cells was due to the lack of NF-_κ_B-mediated gene transcription. However, whether this is the case or whether NEMO possesses additional mechanisms to inhibit apoptosis has not been directly examined. We, therefore, examined more closely the apoptosis induced by TNF in the NEMO-deficient Jurkat T-cell mutant 8321 that we previously isolated.5, 7 This study now demonstrates that NEMO has another antiapoptotic function, which is independent of its role in NF-_κ_B signaling. Early during TNF signaling, NEMO inhibits the activation of the apoptosis pathway by restraining RIP1 from interacting with CASPASE-8.
Results
NEMO-deficient cells are more sensitive to TNF-induced cell-death independent of NF-_κ_B
We have previously reported that NEMO-deficient Jurkat T cells (clone 8321) are highly sensitive to TNF-induced cell death when compared to wild-type cells and this phenotype in the mutant clone can be reverted on complementation with a NEMO cDNA.5 In this study we compared the cell-death sensitivity of the NEMO-deficient cells to that of wild-type parental cells rendered sensitive to apoptosis by stable transfection of the I_κ_B_α_ super-repressor (I_κ_B_α_SR), a potent inhibitor of NF-_κ_B nuclear translocation. As expected, increased cell death was observed in the 3T8 cells transfected with I_κ_B_α_SR (3T8/I_κ_B_α_SR) compared to the 3T8 cells transfected with bacterial GST as an negative control (3T8/Con) at 4 and 8 h after TNF stimulation, consistent with published observations that NF-_κ_B blockade renders cells sensitive to TNF-mediated apoptosis. What was surprising was the observation that NEMO-deficient 8321 cells displayed greater sensitivity to cell death when compared to the 3T8/I_κ_B_α_SR cells at both time points (Figure 1a). As the 8321 and the 3T8/I_κ_B_α_SR cell lines are both devoid of NF-_κ_B signaling, the greater sensitivity of the NEMO-deficient 8321 cell line to death suggested that NEMO may provide additional prosurvival signals independent of the NF-_κ_B signal.
Figure 1
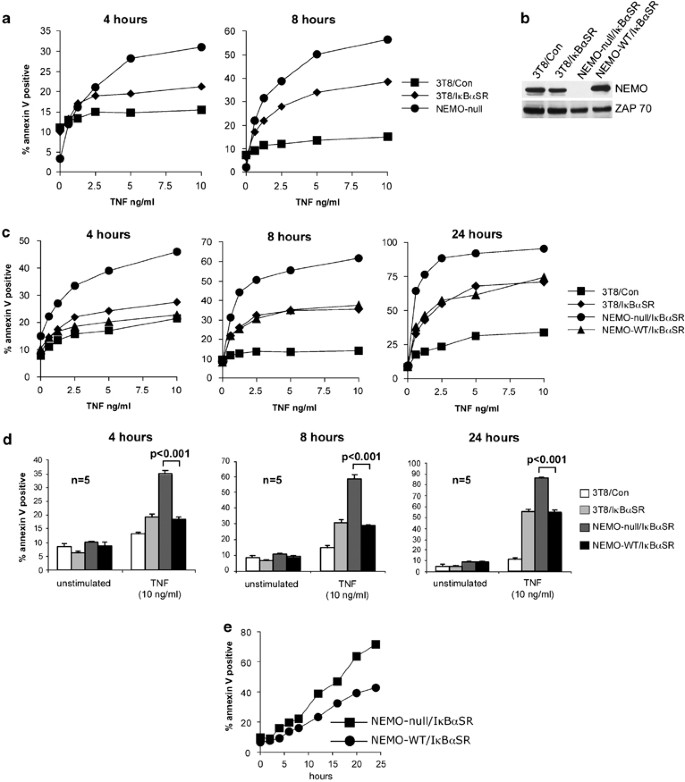
NEMO inhibits TNF-induced apoptosis in a NF-_κ_B-independent manner. (a) Parental 3T8 Jurkat T cells transduced with retrovirus encoding either control protein or I_κ_B_α_SR, and untransfected NEMO-null 8321 cells were treated with the indicated doses of TNF for 4 and 8 h. Cell death was measured by staining with PE-conjugated annexin V and FACS analysis. (b) Cell lysates from 3T8/Con, 3T8/I_κ_B_α_SR, NEMO-null/I_κ_B_α_SR, or NEMO-WT/I_κ_B_α_SR Jurkat T cells were subject to western blot analysis using anti-NEMO followed by anti-ZAP70 as a loading control. (c) 3T8 cells transfected with control protein or I_κ_B_α_SR, and NEMO-null or NEMO-WT Jurkat T cells transfected with I_κ_B_α_SR were treated with the indicated doses of TNF for 4, 8, and 24 h. Cell death was analyzed by staining with PE-conjugated annexin V. The experiments shown are representative of three experiments that were conducted. (d) Cell death was analyzed as in (a) after stimulation with 10 ng/ml of TNF for 4, 8, and 24 h. Each bar indicates the mean±S.D. of quintuplicate samples treated and analyzed independently. _t_-Test was used to calculate P values, and significance was determined at P<0.001. (e) Cells were treated with 5 ng/ml of TNF for the indicated times. Cell death was measured by staining with PE-conjugated annexin V
To test the above hypothesis, the 8321 cell line was reconstituted with either a control protein (designated NEMO-null) or wild-type NEMO (designated NEMO-WT). The two cell lines were further transfected with I_κ_B_α_SR (designated NEMO-null/I_κ_B_α_SR and NEMO-WT/I_κ_B_α_SR) to test the NF-_κ_B-independent prosurvival function of NEMO. Wild-type Jurkat T cells expressing bacterial GST (3T8/Con) or I_κ_B_α_SR (3T8/I_κ_B_α_SR) were included in the analysis as controls. Western blot analysis was performed to confirm that there was equivalent expression of NEMO in the 3T8/Con, 3T8/I_κ_B_α_SR, and NEMO-WT/I_κ_B_α_SR cell lines (Figure 1b). All these cell lines also possess an integrated NF-_κ_B-Thy1 reporter, which allowed us to verify the ability of I_κ_B_α_SR to block NF-_κ_B-mediated gene transcription. On TNF treatment, 3T8/Con expressed Thy1 as a result of NF-_κ_B activation, whereas the three cell lines transfected with I_κ_B_α_SR did not express Thy1 (Supplementary Figure 1), thus confirming that I_κ_B_α_SR does indeed block the NF-_κ_B pathway. These cell lines were treated with increasing concentrations of TNF for 4, 8, and 24 h, and cell death was examined by annexin V staining. Both the 3T8/I_κ_B_α_SR and NEMO-WT/I_κ_B_α_SR cell lines exhibited enhanced sensitivity to TNF-induced cell death when compared to 3T8/Con (Figure 1c). The effect of NF-_κ_B blockade on cell death was minimal at 4 h of TNF treatment and it became more discernable at 8 and 24 h. However, what was striking at the 4 h time point was the noticeably enhanced cell death in NEMO-null/I_κ_B_α_SR cells compared to NEMO-WT/I_κ_B_α_SR cells (Figure 1c and d). This dose-dependent enhancement in cell death was observed at 4 h, and persisted for up to 24 h (Figure 1c and d). To further confirm these results, the NEMO-null/I_κ_B_α_SR and NEMO-WT/I_κ_B_α_SR cell lines were treated with TNF for varying lengths of time up to 24 h. NEMO-null/I_κ_B_α_SR cells undergo more rapid TNF-induced cell death than NEMO-WT/I_κ_B_α_SR cells (Figure 1e). As the two cell lines were both transfected with I_κ_B_α_SR and thus deficient in NF-_κ_B-mediated gene transcription, we conclude that NEMO inhibits TNF-induced cell death independently of NF-_κ_B. Furthermore, the NEMO-deficient cells displayed faster kinetics in their cell death compared to ‘NF-_κ_B-deficient’ cells. These results suggest that in addition to its prosurvival function via the induction of NF-_κ_B-dependent gene transcription, NEMO exerts another inhibitory effect on cell death during TNF signaling before NF-_κ_B-dependent gene transcription.
NEMO-deficient cells undergo more caspase activation than NEMO-sufficient cells
We next examined whether the effect of NEMO deficiency on cell death was due to enhanced activation of the caspase cascade. NEMO-null/I_κ_B_α_SR and NEMO-WT/I_κ_B_α_SR cell lines were treated with varying doses of TNF for 2 and 4 h. Western blot analysis of the cleaved forms of CASPASE-8 (p43/p41 and p18 fragments), CASPASE-3 (p17/p19 fragments), and PARP revealed that NEMO-null/I_κ_B_α_SR cells exhibited greater activation of the caspase cascade when compared to the NEMO-WT/I_κ_B_α_SR cell line (Figure 2a and b). A time-course experiment also revealed a more rapid increase in CASPASE-8, CASPASE-3, and PARP cleavage in NEMO-null/I_κ_B_α_SR cells (Figure 2c). These results indicate that an increase in caspase activation is responsible for the enhancement in cell death observed in NEMO-deficient Jurkat T cells.
Figure 2
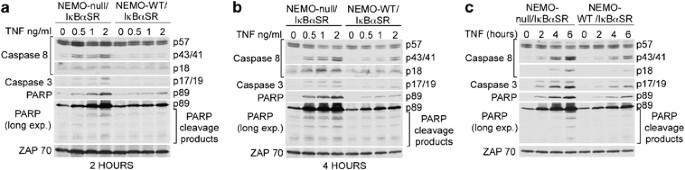
NEMO-deficient cells undergo more caspase activation than NEMO-sufficient cells. (a, b) NEMO-null/I_κ_B_α_SR or NEMO-WT/I_κ_B_α_SR cells were treated with indicated doses of TNF for 2 and 4 h (a and b, respectively). SDS-soluble cell extracts were subject to western blot analysis using antibodies specific to CASPASE-8, CASPASE-3, PARP, and ZAP70 (loading control). The experiments shown are representative of three experiments that were conducted. (c) Cell lines were treated with 2 ng/ml of TNF for the indicated times. SDS-soluble cell extracts were subject to western blot analysis using antibodies specific to CASPASE-8, CASPASE-3, PARP, and ZAP70. The experiments shown are representative of three experiments that were conducted
TNF-induced apoptosis in NEMO-deficient cells is dependent on RIP1
We next determined the NF-_κ_B-independent mechanism that could account for the enhanced cell death in the NEMO-deficient cells. NEMO can bind directly to RIP1 through at least two different domains. First, NEMO can bind constitutively to RIP1 as evidenced by their interactions in yeast two-hybrid analysis8, 9 and this occurs via a pseudo-_α_-helix present in residues 194–260 of NEMO.10 Second, NEMO was recently shown to further bind to RIP1 that has undergone K63-linked polyubiquitination after TNF stimulation11, 12 via an ubiquitin-binding domain present on residues 292–322 of NEMO. We also found that RIP1 serves as a potent death signaling molecule in TNFR1 signaling if it is not ubiquitinated because it is able to bind and rapidly activate CASPASE-8.13 Based on these observations, we hypothesized that one function of NEMO may be to ‘restrain’ RIP1 from engaging CASPASE-8 and, therefore, in NEMO-deficient cells RIP1 is not restrained and it is able to rapidly activate the caspase cascade. This hypothesis predicts that TNF-mediated apoptosis in the NEMO-deficient cells is RIP1-dependent whereas in NEMO-sufficient cells, RIP1 is not involved in apoptosis. To test this, we knocked down RIP1 expression in NEMO-null/I_κ_B_α_SR cell lines using a vector encoding a nonsilencing short hairpin RNA (si-NS) or one targeting RIP1 (si-RIP1). The efficiency of RIP1 knockdown was confirmed by western blot analysis (Figure 3a). Consistent with our hypothesis, there is significantly less apoptosis in NEMO-null/I_κ_B_α_SR cells on RIP1 knockdown (Figure 3b, open circles versus closed circles; Figure 3c, dark gray bars versus black bars). These results demonstrate that TNF-induced apoptosis in NEMO-deficient cells requires RIP1. We also tested the effect of RIP1 knockdown on TNF-induced apoptosis in the 3T8/I_κ_B_α_SR cells. Despite a similar reduction in RIP1 expression (Figure 3a), RIP1 knockdown had no significant effect on apoptosis in the 3T8/I_κ_B_α_SR cells (Figure 3b, open squares versus closed squares; 3C, white bars versus light gray bars). Thus as predicted by our hypothesis, cells with wild-type expression of NEMO that are rendered sensitive to TNF-induced apoptosis by NF-_κ_B blockade (i.e., ‘NF-_κ_B-deficient’ cells) do not require RIP1 to undergo apoptosis. In contrast, NEMO-deficient cells require RIP1 to undergo TNF-induced apoptosis. This demonstrates that the TNF cell-death pathway is mechanistically different in cells that are ‘NF-_κ_B-deficient’ versus cells that are NEMO-deficient.
Figure 3
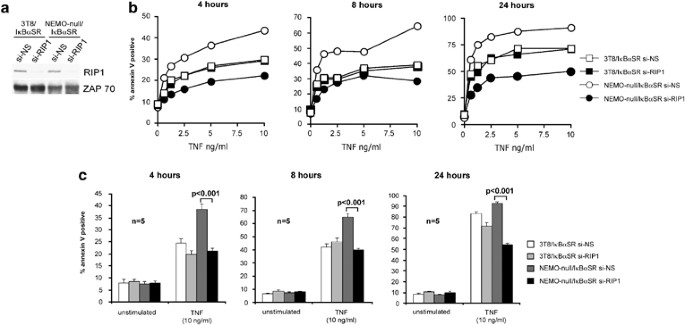
TNF-induced cell death in NEMO-deficient cells requires RIP1. (a) 3T8/I_κ_B_α_SR and NEMO-null/I_κ_B_α_SR cells were infected with retroviruses generated with pSUPERretro-GFP vector encoding shRNA directed against nonsilencing target (si-NS) or RIP1 (si-RIP1). Transduced cells were selected by cell sorting for high GFP expression. Knockdown of RIP1 expression was determined by western blot analysis for RIP1. Anti-ZAP70 blotting was performed as a loading control. (b) The four cell lines used in (a) were treated with the indicated doses of TNF for 4, 8, and 24 h. Cell death was measured by staining with PE-conjugated annexin V and FACS analysis. The experiments shown are representative of three experiments that were conducted. (c) Cell death was analyzed as in (b) after stimulation with 10 ng/ml of TNF for 4, 8, and 24 h. Each bar represents the mean±S.D. of quintuplicate samples treated and analyzed independently. P values <0.001 are indicated
Gene knockout studies have shown the Fas signaling adapter Fadd to be critical in apoptosis in mouse fibroblasts triggered by a combination of TNF and cycloheximide.14 To examine whether FADD plays a role in the TNF-induced cell death observed in NEMO-deficient cells, we knocked down FADD expression in NEMO-null/I_κ_B_α_SR cells using a lentiviral vector encoding a short hairpin RNA targeting FADD (si-FADD) and as a negative control, a nonsilencing hairpin (si-NS). The efficiency of FADD silencing was confirmed by western blot analysis (Supplementary Figure 2A). Silencing of FADD had no effect on TNF-induced apoptosis in the NEMO-null/I_κ_B_α_SR cells. As a positive control, we examined FAS-mediated apoptosis in the same cells. As expected, FADD silencing resulted in decreased sensitivity to FAS-induced cell death demonstrating the effectiveness of the FADD silencing (Supplementary Figure 2B and C). These results suggest that FADD has no role in TNFR1-mediated apoptosis in the NEMO-deficient cells. Recent studies have also reported that FADD knockdown had no effect on cell death caused by TNF plus cycloheximide15 or TNF plus SMAC mimetics.16
NEMO requires its ubiquitin-binding domain for its NF-_κ_B-independent antiapoptotic function
Recent studies have demonstrated that NEMO contains an ubiquitin-binding domain that is able to bind to K63-linked polyubiquitin chains on RIP1.11, 12 Another prediction of the model that NEMO restrains RIP1 is that mutations in NEMO that compromise its binding to ubiquitinated RIP1 will also lead to enhanced sensitivity to apoptosis in an NF-_κ_B-independent manner. To test this prediction, we generated three FLAG-tagged NEMO constructs that contain three substitution mutations (NEMO-Y308S, NEMO-F312A, NEMO-L329P) within the ubiquitin-binding domain that have been shown to abrogate the ability of NEMO to bind to K63-linked polyubiquitin chains.11, 12 NEMO-deficient Jurkat T cells that stably express I_κ_B_α_SR were then reconstituted with a control protein, FLAG NEMO-WT, FLAG NEMO-Y308S, FLAG NEMO-F312A, and FLAG NEMO-L329P. Expression of wild-type NEMO and its mutations were confirmed by western blot analysis (Figure 4a). The absence of NF-_κ_B-mediated gene transcription in all cell lines was confirmed by detection of an NF-_κ_B-Thy1 reporter (Supplementary Figure 3). We treated the NEMO-null/I_κ_B_α_SR, NEMO-WT/I_κ_B_α_SR, NEMO-Y308S/I_κ_B_α_SR, NEMO-F312A/I_κ_B_α_SR, and NEMO-L329P/I_κ_B_α_SR cell lines with 5 and 10 ng/ml of TNF for 4, 8, and 24 h. As observed previously, reconstitution with NEMO-WT protected cells from TNF-induced apoptosis, independent of NF-_κ_B (Figure 4b). NEMO-F312A/I_κ_B_α_SR and NEMO-L329P/I_κ_B_α_SR exhibited slightly more cell death at 4 h of TNF treatment, as compared to NEMO-WT/I_κ_B_α_SR (Figure 4b). At 8 and 24 h of TNF treatment, cell lines expressing the three NEMO substitution mutations exhibited significantly more death than cells expressing NEMO-WT (Figure 4b). Thus, as predicted, substitution mutations in the ubiquitin-binding region of NEMO enhanced sensitivity to TNF-induced apoptosis in cells devoid of NF-_κ_B-mediated transcription. We then confirmed that the NEMO point mutants were indeed unable to associate with ubiquitinated RIP1 on TNFR1 engagement in the context of I_κ_B_α_SR expression. Consistent with published data,11, 12 only NEMO-WT was able to associate with ubiquitinated RIP1 in TNF-stimulated cells (Figure 4c, lane 4). Therefore, the inability to associate with ubiquitinated RIP1 in the NEMO substitution mutants correlated with enhanced apoptosis. Although all three point mutations within the ubiquitin-binding domain abrogated NEMO's interaction with ubiquitinated RIP1 to a similar degree (Figure 4c), there appears to be some small differences in their ability to block cell death (Figure 4b). It is unclear why we did not observe a complete correlation between binding to ubiquitinated RIP1 and blockade of cell death. One possibility could be that the cell-death assay is more sensitive and quantitative than the ‘RIP1-laddering’ analysis in the NEMO-RIP1 co-immunoprecipitations. Therefore, subtle differences in these mutants may not be discernable by the ‘RIP1-laddering’ assay in Figure 4c. Alternatively, the three mutations may have different affects on binding to additional partner proteins involved in the regulation of this response that have yet to be characterized. NEMO mutants defective in ubiquitin binding were still able to associate with unmodified RIP1 (Figures 4c, lanes 3–10), consistent with reports that NEMO and RIP1 can also associate independently of ubiquitination as evidenced by their ability to associate in yeast two-hybrid analysis.8, 9 This ubiquitin-independent association could account for the faster kinetics of cell death in the NEMO-null cells compared to cells reconstituted with the NEMO ubiquitin-binding mutants (Figure 4b). Taken together, these results support the model that NEMO sequesters and restrains RIP1 from activating caspases.
Figure 4
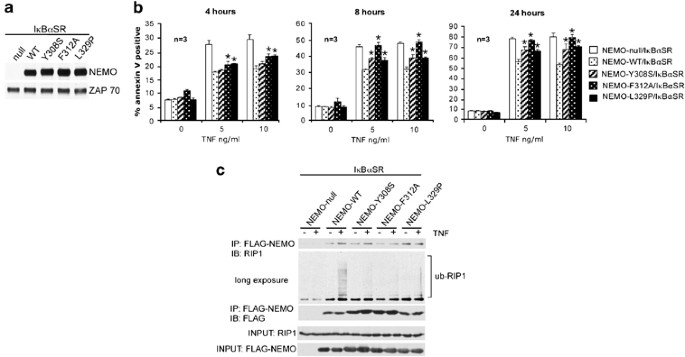
NEMO requires its ubiquitin-binding domain for its NF-_κ_B-independent antiapoptotic function. (a) Cell lysates from NEMO-null/I_κ_B_α_SR, NEMO-WT/I_κ_B_α_SR, NEMO-Y308S/I_κ_B_α_SR, NEMO-F312A/I_κ_B_α_SR, NEMO-L329P/I_κ_B_α_SR cells were subject to western blot analysis using anti-NEMO followed by anti-ZAP70 as a loading control. (b) Cell lines from (a) were treated with 5 or 10 ng/ml of TNF for 4, 8, and 24 h. Cell death was measured by staining with PE-conjugated annexin V and FACS analysis. Each bar represents the mean±S.D. of triplicate samples treated and analyzed independently. _t_-Test was used to calculate P values and asterisks indicate P<0.01 when compared to values from the NEMO-WT/I_κ_B_α_SR cell line. (c) Cell lines from (a) were treated with 100 ng/ml of TNF for 15 min. FLAG-tagged wild-type NEMO or NEMO mutants were immunoprecipitated from total cell lysates using anti-FLAG M2 affinity beads. The immunoprecipitates were sequentially blotted with anti-RIP1 (first and second panels) and anti-FLAG (third panel) antibodies. Blotting with the two antibodies on 1% of the total cell extracts are shown in the bottom two panels. The experiment shown is representative of three experiments that were conducted
NEMO inhibits apoptosis by restraining RIP1 from associating with CASPASE-8
Another prediction of the model is that in NEMO-sufficient cells, RIP1 will not form a signaling complex with CASPASE-8 whereas in NEMO-deficient cells, RIP1 rapidly associates with CASPASE-8. Lysates from NEMO-null and NEMO-WT cells treated with TNF for 1 and 2 h were subjected to immunoprecipitation using an antibody to RIP1 followed by blotting with an antibody that recognizes active CASPASE-8. We used the rabbit monoclonal 18C8, which only recognizes active cleaved CASPASE-8 migrating at 43/41 and 18 kD, because PROCASPASE-8 migrated at the same mobility as the immunoglobulin heavy chain from the RIP1 immunoprecipitates. The presence of the heavy chain precluded us from detecting the uncleaved PROCASPASE-8 in western blots. As predicted, we detected RIP1 association with active CASPASE-8 in NEMO-null cells but not in the NEMO-WT cells (Figure 5a). As the experiment shown in Figure 5a was conducted using cells without NF-_κ_B blockade, we repeated the same experiment in cells that were also transfected with I_κ_B_α_SR to confirm that the effect of NEMO on the interaction between RIP1 and CASPASE-8 was indeed NF-_κ_B-independent. As shown in Figure 5b, the result from cells transfected with I_κ_B_α_SR was similar to that observed in cells that were not transfected with I_κ_B_α_SR. There was more active CASPASE-8 associated with RIP1 in the NEMO-null/I_κ_B_α_SR cells compared to the NEMO-WT/I_κ_B_α_SR cells. This demonstrates that NEMO inhibits the association of RIP1 and CASPASE-8 independent of its role in NF-_κ_B. The RIP1 immunoprecipitates were reprobed with anti-FADD to examine whether NEMO had any effect on the association of RIP1 and FADD. Interestingly, whereas there were no discernable changes in RIP1–FADD interaction in response to TNF stimulation in the NEMO-null/I_κ_B_α_SR cells, FADD appeared to disassociate from RIP1 in the NEMO-WT/I_κ_B_α_SR cells. This suggests that the absence of NEMO stabilizes the RIP1–FADD interaction and the resultant higher level of FADD in the complex may potentially contribute toward enhancing the interaction between RIP1 and CASPASE-8. Nonetheless, the fact that FADD knockdown had no effect on TNF-induced apoptosis in the NEMO-deficient cells (Supplementary Figure 2) argues against a significant role for FADD in this pathway.
Figure 5
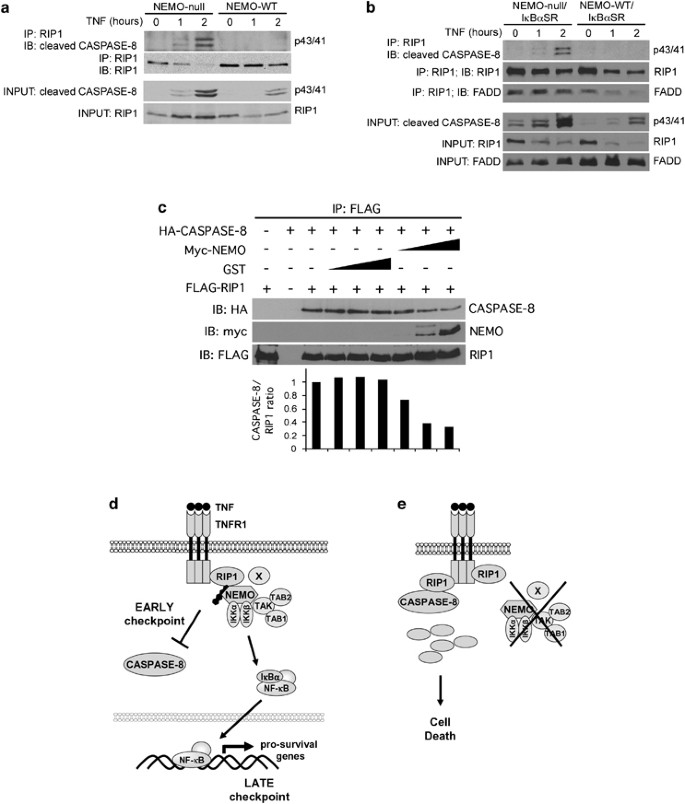
NEMO inhibits RIP1 from associating with CASPASE-8. (a) NEMO-null and NEMO-WT Jurkat T cells were treated with 25 ng/ml TNF for 1 and 2 h. RIP1 was immunoprecipitated from the lysates using anti-RIP1 antibody. The protein complexes were eluted and subject to sequential western blot analysis using anticleaved CASPASE-8 and anti-RIP1 antibodies. (b) NEMO-null/I_κ_B_α_SR and NEMO-WT/I_κ_B_α_SR cells were treated as in panel (a). Blotting was performed sequentially with anticleaved CASPASE-8, anti-FADD, and anti-RIP1 antibodies. (c) 293EBNA cells were transfected with FLAG-RIP1, and HA-CASPASE-8 in the presence of increasing amounts of myc-NEMO or the negative control GST. Anti-FLAG immunoprecipitations were eluted with the FLAG peptide and sequentially blotted with anti-HA, anti-myc, and anti-FLAG. The intensities of the CASPASE-8 bands were determined by densitometry and normalized to the corresponding RIP1 band. The ratios of the intensity in the different samples are presented relative to the ratio in the sample with no GST or NEMO. (d) Model of TNFR1 signaling showing two cell-death checkpoints. On ligation of TNFR1, ubiquitination of RIP1 occurs rapidly leading to its association with NEMO and other components of the IKK signaolsome. This sequesters RIP1 away from CASPASE-8 and serves as the early transcription-independent cell-death checkpoint. The formation of the IKK signalosome subsequently leads to the induction of NF-_κ_B-dependent antiapoptotic genes and this serves as a second cell-death checkpoint, which provides for a long-lasting genetically programmed protection from death. (e) If the early cell-death checkpoint fails, such as when ubiquitination of RIP1 is prevented or if a regulatory component such as NEMO is absent, RIP1 can engage CASPASE-8 and death rapidly ensues. This early NF-_κ_B-independent cell-death checkpoint serves to determine whether the survival or death pathway is triggered following TNFR1 ligation
Finally, we tested whether NEMO can function to inhibit the interaction between RIP1 and CASPASE-8 in cotransfection experiments. Equivalent amounts of FLAG-RIP1 and HA-CASPASE-8 were cotransfected into 293EBNA cells in the presence of increasing amounts of NEMO or the negative control GST. RIP1 was immunoprecipitated using anti-FLAG mAb, eluted with the FLAG peptide, and blotted for the presence of HA-CASPASE-8. As shown in Figure 5c, the presence of NEMO reduced the amount of CASPASE-8 associated with RIP1 suggesting that NEMO can disrupt the association of RIP1 with CASPASE-8. This observation provides further evidence to support the model that NEMO acts to restrain RIP1 from associating with CASPASE-8, thus preventing cell death.
Discussion
NEMO-deficient cells are sensitive to TNF-induced apoptosis4, 5, 6 and as NEMO has been shown to be necessary for NF-_κ_B activation,3, 17 it has been assumed that NEMO functioned as an antiapoptotic molecule due to its role in the NF-_κ_B signaling pathway. However, this study demonstrates an additional, novel role for NEMO in blocking cell death independent of its role in NF-_κ_B signaling whereby NEMO restrains RIP1 from engaging CASPASE-8 before the induction of prosurvival genes. In addition to its well-characterized role in TNFR1-to-NF-_κ_B signaling,11, 13, 18, 19, 20 RIP1 is also known to be a potent cell-death inducer.21, 22 Therefore, it is likely that cells expend considerable efforts to rein in the apoptosis-inducing behavior of RIP1 when death is not the desired outcome. Based on this study and our previous study,13 at the minimum, this consists of ubiquitination of RIP1 and sequestration of RIP1 by NEMO away from CASPASE-8. It is likely that E3 ligases such as TRAF213 and CIAPs16, 23 and deubiquitinases such as CYLD16 may have a role in regulating this process.
Although the focus in recent years has been on NF-_κ_B as the determinant of survival versus death following TNF stimulation, our molecular characterization of an early cell-death checkpoint that is NF-_κ_B-independent suggests that the current model of TNF signaling should be revised to incorporate this new understanding. We, therefore, propose that there are two cell-death checkpoints during TNF signaling. An early checkpoint occurs when RIP1 undergoes ubiquitination mediated by TRAF2 or c-IAPs16, 23, 24 and this enhances its association with NEMO and restrains RIP1 from engaging CASPASE-8 (Figure 5d). The main function of this first checkpoint is to ensure that RIP1 does not trigger the caspase cascade. At the same time, formation of the RIP1–NEMO complex also leads to IKK activation and this leads to a later cell-death checkpoint in which NF-_κ_B now induces prosurvival gene expression, which now provides for a long lasting protection from cell death. At the first cell-death checkpoint, if ubiquitination of RIP1 is prevented or if a regulatory component such as NEMO is absent, RIP1 is now free to engage CASPASE-8 and death rapidly ensues (Figure 5e). This model, which shows that there are two cell-death checkpoints during TNF signaling provides an explanation for the paradoxical observations that the TNF death signaling machinery is preexisting in cells whereas the survival response is dependent on transcription, yet cells are largely resistant to TNF-induced cell death. Soon after TNFR1 is ligated, RIP1 is rapidly ubiquitinated and sequestered from preexisting components of the death pathway such as CASPASE-8. This transcription-independent event then allows sufficient time for the NF-_κ_B transcription machinery to induce the array of prosurvival genes necessary to permanently disable the cell-death pathway. Conversely, if the desired biological outcome is death, disabling the first cell-death checkpoint will render cells susceptible to rapid TNF-mediated cell death. Physiologically, this may be achieved by altering the levels of ubiquitin-modifying enzymes of RIP1 including E3 ligases and deubiquitinases. These ubiquitin-modifying enzymes, as well as nonenzymatic molecules such as NEMO that regulate the interaction of RIP1 with the caspase pathway, are attractive targets for pharmacological modulation. In this regard, two recent studies reported that SMAC mimetics, which cause the autodegradation of CIAP1 and CIAP2, render tumor cell lines sensitive to TNF-mediated apoptosis through a RIP1-dependent manner.16, 23 In the absence of both CIAP1 and CIAP2, RIP1 does not undergo ubiquitination and converts to a death signaling molecule, an effect analogous to the loss of the ubiquitin acceptor site lysine 377 on RIP1 we previously described.13 Therefore, it seems likely that the loss of K377 on RIP1,13 inhibition of TRAF2,13 loss of CIAP1 and CIAP2 caused by SMAC mimetics,16, 23 and the loss of NEMO described in this study all function to disrupt the early NF-_κ_B-independent cell-death checkpoint in TNF signaling.
It is very likely that disruption of the first cell-death checkpoint generates a qualitatively and quantitatively different response to TNF-induced cell death when compared to disruption of the second cell-death checkpoint. Death sensitivity conferred by the loss of the first checkpoint is dependent on RIP1 and is a direct effect of the formation of the signaling complex between RIP1 and CASPASE-8 (Figure 3b, open circles versus closed circles). In contrast, death sensitivity conferred by loss of the second checkpoint does not require RIP1 and is an indirect effect caused by the loss of caspase inhibitors such as c-FLIP secondary to the loss of NF-_κ_B (Figure 3b, open squares versus closed squares). The faster kinetics and greater magnitude of death in the NEMO-deficient cells, which has a disruption in both the first and second checkpoint, as compared to the I_κ_B_α_SR-transfected cells, which has a disruption in the second checkpoint, indicated that disruption of the first checkpoint generates a more potent death signal.
The observation that nonubiquitinated RIP1 forms a complex with CASPASE-8 to trigger apoptosis13, 16, 23 raises a number of questions. For instance, a vast proportion of RIP1 in wild-type cells does not undergo ubiquitination after TNF stimulation and yet they do not associate with CASPASE-8 to trigger apoptosis. Therefore, it is likely that preventative mechanisms must be in place in quiescent cells to limit these potentially deadly associations from occurring. These preventative mechanisms are poorly understood and may involve (1) keeping the localization of these molecules apart, (2) stimulus-dependent posttranslational modifications needed for the associations to occur, and (3) inhibitors bound to RIP1 or CASPASE-8 that prevent them from interacting. It is potentially interesting that NEMO and RIP1 can interact with each other in yeast8, 9 and in nonstimulated cells (see Figure 4c) leading to the speculation that under basal condition, binding of NEMO to RIP1 may serve to sequester RIP1 from CASPASE-8. The observation in Figure 5c that transfection of NEMO displaces CASPASE-8 from RIP1 is also consistent with this speculation. Future experiments will be directed at testing which of these preventative mechanisms may be involved in the TNF pathway. Exactly how NEMO binding to ubiquitinated RIP1 prevents RIP1 from associating with CASPASE-8 remains to be discerned. Binding of ubiquitinated RIP1 to NEMO may result in RIP1's localization to a cellular compartment that is not accessible to CASPASE-8. In addition, NEMO binding to ubiquitinated RIP1 may also serve to stabilize this posttranslational modification by preventing deubiquitinases such as A20 and CYLD from gaining access to the polyubiquitin chains on RIP1. Whether the attachment or removal of polyubiquitin chains on RIP1 is altered in the NEMO-deficient cells is unclear at the moment. It is possible that in the NEMO-deficient cells, ubiquitination of RIP1 is normal but due to the lack of binding to NEMO, the ubiquitinated RIP1 can now form a complex with CASPASE-8. These possibilities will be tested in the future.
We also examined whether FADD had any role in the cell-death process following disruption of the first cell-death checkpoint caused by the loss of NEMO. Although the loss of NEMO appeared to stabilize the RIP1–FADD interaction (Figure 5b), suggesting FADD may play a role here, knocking down FADD had no effect on the overall cell death triggered by TNF (Supplementary Figure 2) arguing against a role for FADD. Nonetheless, we are unable to rule out that in the absence of FADD, there may be a qualitative change in the cell death that is occurring. Harper et al.25 previously reported that FADD deficiency in Jurkat T cells led to increased TNF-mediated necrosis. It is possible that in our NEMO-deficient cells, FADD silencing shifted the death response from apoptosis to necrosis, which we are unable to distinguish by annexin V staining. We are currently exploring whether nonapoptotic cell death may occur in the NEMO-deficient cells and whether FADD and RIP1 have any role in it.
Although this study demonstrates that NEMO plays a prosurvival role in TNF signaling through multiple mechanisms, there is one intriguing observation that suggests perhaps NEMO may have a proapoptotic role as well. In cells that are RIP1 deficient and NF-_κ_B deficient, the absence of NEMO leads to less cell death (Figure 3b, black squares versus black circles) suggesting that under these conditions, NEMO is proapoptotic. The biological relevance of this putative proapoptotic function of NEMO is unclear at the moment because there are currently no reports of RIP1 deficiency in cells other than that achieved by experimental mutagenesis or knockdown. One possibility may be that because RIP1 and NEMO are binding partners, the loss of RIP1 may allow NEMO to now bind to another molecule that is strongly proapoptotic. Future studies will be directed at examining this putative proapoptotic function of NEMO.
TNF is a dual-functioning molecule in that it can promote cell survival or cell death. Cells are able to fine-tune the balance between these two opposing responses to generate the appropriate response. Understanding the mechanism underlying this balance between survival and death may suggest strategies to manipulate this balance for therapeutic purposes in diseases where TNF may have a role such as cancer, inflammatory bowel disease, and rheumatoid arthritis. Our results suggest that disrupting the NEMO–RIP1 interaction, that is, the first cell-death checkpoint in the TNFR1 pathway, may be one such potential strategy.
Materials and Methods
Cell lines
The wild-type and NEMO/IKK_γ_-deficient Jurkat T cell lines have been described previously.5 Cells were maintained in IMDM medium (Sigma-Aldrich) supplemented with 10% bovine calf serum (Hyclone), 50 mM 2-mercaptoethanol (Sigma-Aldrich), 2 mM L-glutamine (Cellgro), and 15 _μ_g/ml gentamicin. Retroviral transduction of Jurkat T cells was performed as previously described.5
Plasmids
I_κ_B_α_SR, which contains a serine to alanine substitution mutation at residues 32 and 36, was subcloned into a retroviral expression vector containing an internal ribosome entry site (IRES)-puromycin resistance cassette. Bacterial glutathione S_-transferase (GST) cloned into the same retroviral vector was used as a negative control. Bulk populations of puromycin-resistant cells were used in all experiments. Wild-type NEMO/IKK_γ containing a FLAG-tag at the N terminus was subcloned into the retroviral expression vector. NEMO substitution mutations tyrosine to serine at residue 308 (NEMO-Y308S), phenylalanine to alanine at residue 312 (NEMO-F312A), and leucine to proline at residue 329 (NEMO-L329P) were generated using Stratagene Quikchange XL site-directed mutagenesis kit.
RNA interference
ShRNA-mediated gene silencing of RIP1 was performed by using the retroviral expression vector pSUPERretro (Oligoengine) that was modified so that the puromycin-resistance cassette was replaced by green fluorescent protein (GFP). Selection for retrovirus-infected cells expressing high levels of short-hairpin RNA (shRNA) was achieved by sorting for cells with high GFP expression using FACSVantage Cell Sorter (BD Biosciences). The RIP1-specific insert consisted of a 19-nt sequence (GGAGCAAACTGAATAATGA) separated by a noncomplementary spacer from the reverse complement of the same 19-nt sequence to form the shRNA duplex (si-RIP1). Vector containing a nonsilencing shRNA (TTCTCCGAACGTGTCACGT) was used as a control (si-NS).
ShRNA-mediated gene silencing of FADD was performed using the lentivirus vector pLKO.1 containing scramble shRNA (scramble; Addgene Inc., Cambridge, MA, USA) or shRNA targeting FADD (si-FADD) (Sigma-Aldrich). Lentiviral transduction of Jurkat T cells were performed using VSV-G and psPAX2 (Addgene Inc.) expression plasmids for packaging the lentiviral particles and viral infection carried out as previously described for retrovirus.5 Bulk population of drug-resistant cells was used in experiments to avoid clonal variability.
Immunoblotting
Cells were treated as indicated, pelleted, and lysed in buffer containing 20 mM Tris pH 7.5, 40 mM sodium chloride, 5 mM EDTA, 50 mM sodium fluoride, 30 mM sodium pyrophosphate, 1% Triton X-100, and Protease Inhibitor Cocktail Set V (EMD Biosciences Inc.) for 10 min on ice. Lysates were clarified by centrifugation at 10 000 × g at 4°C. Extracts were resolved by SDS-PAGE, transferred onto nitrocellulose membrane, and probed with the indicated antibody and the appropriate secondary antibody conjugated to horseradish peroxidase (Dako Cytomation). Anti-NEMO/IKK_γ_ was obtained from Santa Cruz Biotechnology. Anti-RIP1 antibody was obtained from BD Biosciences. ZAP70 antibody was a generous gift from Dr. Paul Leibson. To analyze the apoptosis pathway, cells were treated with human TNF_α_ (R&D Systems) as indicated. Cells were washed with PBS and lysed in 0.5% SDS by vortexing and boiling for 5 min. Lysates were resolved by SDS-PAGE, transferred onto nitrocellulose membrane, and probed with the indicated antibodies. Caspase 8, cleaved-caspase3, and PARP antibodies were obtained from Cell Signaling Technology.
Flow cytometry
Cells were treated with human TNF_α_ as indicated and stained with either phycoerythrin (PE)-conjugated annexin V (BD Biosciences), or anti-Thy1 (Sera Laboratories) followed by PE-conjugated goat anti-mouse Ig (BD Biosciences). Analysis was performed using a FACScan or FACScalibur flow cytometer and Cell Quest software (Becton Dickinson).
Immunoprecipitation
For FLAG immunoprecipitation, cells were stimulated with TNF as indicated and lysed in buffer containing 20 mM Tris pH 7.5, 150 mM sodium chloride, 1 mM EDTA, 30 mM sodium fluoride, 2 mM sodium pyrophosphate, 5 mM N-ethylmaleimide, 1% Triton X-100, and Protease Inhibitor Cocktail Set V for 10 min on ice.12 Lysates were clarified by centrifugation at 10 000 × g at 4°C and precleared with albumin-agarose beads (Sigma-Aldrich) with agitation for 1 h at 4°C. The beads were removed by centrifugation at 8000 × g for 30 s and the lysate was incubated with anti-FLAG M2 agarose affinity beads (Sigma-Aldrich) with agitation for 2 h at 4°C. Beads were collected by centrifugation at 8000 × g for 30 s and washed five times with lysis buffer containing 500 mM NaCl. Protein was eluted from beads by competition with 300 _μ_g/ml of FLAG peptide (Sigma-Aldrich) in sample buffer. Lysates were resolved by SDS-PAGE and analyzed using anti-RIP1 and anti-FLAG (Sigma-Aldrich) antibodies. RIP1 immunoprecipitation was performed as previously described, with modifications.13 Briefly, cells were stimulated with TNF as indicated and lysed in 0.2% NP-40 lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM sodium chloride, 0.2% NP-40, 10% glycerol, Protease Inhibitor Cocktail Set V, 1 ml/ml BSA) for 15 min on ice. Lysates were clarified by centrifugation at 10 000 × g at 4°C and incubated with albumin-agarose beads (Sigma-Aldrich) with agitation for 1 h at 4°C. The beads were removed by centrifugation at 8000 × g for 30 s, and the lysate was incubated with 2 _μ_g of anti-RIP1 antibody with agitation for 4 h at 4°C followed by incubation with protein A/G agarose beads (Santa Cruz Biotechnology) for 1 h at 4°C. Beads were washed five times with lysis buffer, and proteins were eluted from the beads at 75°C for 10 min. Samples were resolved by SDS-PAGE and analyzed by immunoblotting using a rabbit mAb clone 18C8 that recognizes only cleaved CASPASE-8 (Cell Signaling Technology), anti-FADD (BD Biosciences), and anti-RIP1 antibodies.
Coexpression and coimmunoprecipitation
293EBNA cells were transfected with 2 _μ_g each of plasmids encoding FLAG-RIP1 and HA-CASPASE-8 (kindly provided by Dr. Shahrooz Rabizadeh, Buck Institute for Age Research) in the presence of 0.02, 0.1, and 0.5 _μ_g of myc-NEMO or the negative control GST. zVAD-FMK (10 _μ_M) was added to the transfectants to prevent cell death. Twenty-four hours after transfection, cells were lysed in buffer containing 20 mM Tris pH 7.5, 150 mM sodium chloride, 5 mM EDTA, 50 mM sodium fluoride, 30 mM sodium pyrophosphate, 1% Triton X-100, and Protease Inhibitor Cocktail Set V for 10 min on ice. Lysates were clarified by centrifugation at 10 000 × g at 4°C. FLAG immunoprecipitation was performed as described earlier. Briefly, lysates were precleared with albumin-agarose beads and then incubated with anti-FLAG M2 agarose affinity beads. Protein was eluted from the beads by competition with 300 _μ_g/ml of FLAG peptide. Lysates were resolved by SDS-PAGE and analyzed by western blot using anti-HA (Roche Diagnostics), anti-c-Myc (Santa Cruz Biotechnology), and anti-FLAG (Sigma-Aldrich) antibodies. The intensities of the CASPASE-8 bands were quantified by densitometry analysis and normalized to the corresponding RIP1 bands using the software Quantity One (BioRad).
Statistical analysis
Data are presented as mean±S.E.M. Comparisons between groups were made using Student's _t_-test.
Abbreviations
I_κ_B_α_SR:
I_κ_B_α_ super repressor
IKK_γ_:
I_κ_B kinase-γ
NEMO:
NF-_κ_B essential modifier
RIP1:
receptor interacting protein 1
TNFR1:
TNF receptor 1
shRNA:
short-hairpin RNA
References
- Aggarwal BB . Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 2003; 3: 745–756.
Article CAS Google Scholar - Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Article CAS Google Scholar - Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 1998; 93: 1231–1240.
Article CAS Google Scholar - Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J et al. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev 2000; 14: 854–862.
CAS PubMed PubMed Central Google Scholar - He KL, Ting AT . A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol Cell Biol 2002; 22: 6034–6045.
Article CAS Google Scholar - Makris C, Roberts JL, Karin M . The carboxyl-terminal region of IkappaB kinase gamma (IKKgamma) is required for full IKK activation. Mol Cell Biol 2002; 22: 6573–6581.
Article CAS Google Scholar - He KL, Ting AT . Essential role for IKKgamma/NEMO in TCR-induced IL-2 expression in Jurkat T cells. Eur J Immunol 2003; 33: 1917–1924.
Article CAS Google Scholar - Zhang SQ, Kovalenko A, Cantarella G, Wallach D . Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity 2000; 12: 301–311.
Article CAS Google Scholar - Li Y, Kang J, Friedman J, Tarassishin L, Ye J, Kovalenko A et al. Identification of a cell protein (FIP-3) as a modulator of NF-kappaB activity and as a target of an adenovirus inhibitor of tumor necrosis factor alpha-induced apoptosis. Proc Natl Acad Sci USA 1999; 96: 1042–1047.
Article CAS Google Scholar - Sebban H, Yamaoka S, Courtois G . Posttranslational modifications of NEMO and its partners in NF-kappaB signaling. Trends Cell Biol 2006; 16: 569–577.
Article CAS Google Scholar - Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ . Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22: 245–257.
Article CAS Google Scholar - Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD . Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat Cell Biol 2006; 8: 398–406.
Article CAS Google Scholar - O’Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT . Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol 2007; 17: 418–424.
Article Google Scholar - Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 1998; 279: 1954–1958.
Article CAS Google Scholar - Jin Z, El-Deiry WS . Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol Cell Biol 2006; 26: 8136–8148.
Article CAS Google Scholar - Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Article CAS Google Scholar - Rothwarf DM, Zandi E, Natoli G, Karin M . IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998; 395: 297–300.
Article CAS Google Scholar - Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P . The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998; 8: 297–303.
Article CAS Google Scholar - Li H, Kobayashi M, Blonska M, You Y, Lin X . Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem 2006; 281: 13636–13643.
Article CAS Google Scholar - Ting AT, Pimentel-Muinos FX, Seed B . RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J 1996; 15: 6189–6196.
Article CAS Google Scholar - Pimentel-Muinos FX, Seed B . Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity 1999; 11: 783–793.
Article CAS Google Scholar - Stanger BZ, Leder P, Lee TH, Kim E, Seed B . RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995; 81: 513–523.
Article CAS Google Scholar - Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
Article CAS Google Scholar - Lee TH, Shank J, Cusson N, Kelliher MA . The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem 2004; 279: 33185–33191.
Article CAS Google Scholar - Harper N, Hughes M, MacFarlane M, Cohen GM . Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem 2003; 278: 25534–25541.
Article CAS Google Scholar
Acknowledgements
We thank the Mount Sinai Flow Cytometry Shared Research Facility for assistance with cell sorting, and members of Paul Leibson's lab for ZAP70 antibody. This work was supported by National Institutes of Health grants AI052417 and AI057997, and a grant from the New York Chapter of the Arthritis Foundation to ATT. MAO’D is a recipient of a Research Fellowship Award from the Crohn's and Colitis Foundation of America. DL-A is a recipient of the National Institutes of Health Ruth L Kirschstein NRSA Postdoctoral Award (AI065058). ATT is a recipient of the Irma T Hirschl Career Scientist Award.
Author information
Author notes
- D Legarda-Addison and H Hase: These two authors contributed equally to this work.
Authors and Affiliations
- Immunology Institute, Mount Sinai School of Medicine, New York, 10029, NY, USA
D Legarda-Addison, H Hase, M A O'Donnell & A T Ting - Division of Immunology, Dokkyo Medical University School of Medicine, Tochigi, Japan
H Hase
Authors
- D Legarda-Addison
- H Hase
- M A O'Donnell
- A T Ting
Corresponding author
Correspondence toA T Ting.
Additional information
Edited by P Vandenabeele
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Legarda-Addison, D., Hase, H., O'Donnell, M. et al. NEMO/IKK_γ_ regulates an early NF-_κ_B-independent cell-death checkpoint during TNF signaling.Cell Death Differ 16, 1279–1288 (2009). https://doi.org/10.1038/cdd.2009.41
- Received: 01 July 2008
- Revised: 10 March 2009
- Accepted: 16 March 2009
- Published: 17 April 2009
- Issue Date: September 2009
- DOI: https://doi.org/10.1038/cdd.2009.41