Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012 (original) (raw)
- Review
- Published: 15 July 2011
- I Vitale1,2,3,
- J M Abrams4,
- E S Alnemri5,
- E H Baehrecke6,
- M V Blagosklonny7,
- T M Dawson8,
- V L Dawson8,
- W S El-Deiry9,
- S Fulda10,
- E Gottlieb11,
- D R Green12,
- M O Hengartner13,
- O Kepp1,2,3,
- R A Knight14,
- S Kumar15,16,
- S A Lipton17,18,19,20,
- X Lu21,
- F Madeo22,
- W Malorni23,24,
- P Mehlen25,26,27,28,
- G Nuñez29,
- M E Peter30,
- M Piacentini31,32,
- D C Rubinsztein33,
- Y Shi34,
- H-U Simon35,
- P Vandenabeele36,37,
- E White38,
- J Yuan39,
- B Zhivotovsky40,
- G Melino41,42 &
- …
- G Kroemer1,43,44,45,46
Cell Death & Differentiation volume 19, pages 107–120 (2012)Cite this article
- 38k Accesses
- 1904 Citations
- 25 Altmetric
- Metrics details
Abstract
In 2009, the Nomenclature Committee on Cell Death (NCCD) proposed a set of recommendations for the definition of distinct cell death morphologies and for the appropriate use of cell death-related terminology, including ‘apoptosis’, ‘necrosis’ and ‘mitotic catastrophe’. In view of the substantial progress in the biochemical and genetic exploration of cell death, time has come to switch from morphological to molecular definitions of cell death modalities. Here we propose a functional classification of cell death subroutines that applies to both in vitro and in vivo settings and includes extrinsic apoptosis, caspase-dependent or -independent intrinsic apoptosis, regulated necrosis, autophagic cell death and mitotic catastrophe. Moreover, we discuss the utility of expressions indicating additional cell death modalities. On the basis of the new, revised NCCD classification, cell death subroutines are defined by a series of precise, measurable biochemical features.
Similar content being viewed by others
Preface
Since the first descriptions of programmed cell death mechanisms, which date back to the mid-1960s,1, 2, 3 several attempts have been made to classify cell death subroutines based on morphological characteristics. Thus, in 1973 Schweichel and Merker4 proposed a classification of several cell death modalities, including ‘type I cell death’ associated with heterophagy, ‘type II cell death’ associated with autophagy and ‘type III cell death’, which was not associated with any type of digestion, corresponding to apoptosis, autophagic cell death and necrosis, respectively.5, 6
Even though deep insights into the molecular pathways that regulate and execute cell death have been gained and biochemical assays for monitoring cell death-related phenomena have become laboratory routine, the scientific community has not yet adopted a systematic classification of cell death modalities based on biochemical rather than morphological criteria. Nonetheless, there has been a tendency to dichotomize cell death events into either of two mutually exclusive groups. Thus, caspase-dependent, tolerogenic, programmed and physiological cell death instances have been contrasted to their caspase-independent, immunogenic, accidental and pathological counterparts, respectively.7
The Nomenclature Committee on Cell Death (NCCD) has formulated two subsequent rounds of recommendations in 2005 and 2009, in Cell Death and Differentiation.6, 8 Therein, unified criteria for the definition of cell death morphotypes were proposed and guidelines on the use of cell death-related terminology were given. The mission of the NCCD, as formulated previously, is ‘to provide a forum in which names describing distinct modalities of cell death are critically evaluated and recommendations on their definition and use are formulated, hoping that a non-rigid, yet uniform nomenclature will facilitate the communication among scientists and ultimately accelerate the pace of discovery’.8 In line with this mission statement and following recent breakthroughs in cell death research that have invalidated the notion that necrosis would represent a merely accidental cell death mode (see below),9, 10, 11, 12 the NCCD believes that the time has become appropriate for a novel systematic classification of cell death based on measurable biochemical features.
Pros and Cons of Morphological Versus Biochemical Classifications of Cell Death
The very first catalogs of cell death4 necessarily relied on morphological traits, because the biochemical tests that are available nowadays for assessing the cell demise13 were only developed decades later. Nevertheless, morphological classifications have dominated the cell death research scene even after the introduction of biochemical assays into the laboratory routine. Several economical, methodological, educational and theoretical reasons can be invoked to explain why the scientific community has clung to a conservative, morphological classification of cell death modalities. First, while conventional light microscopy is available in all cell biology laboratories, this is not the case for more sophisticated equipment (e.g., fluorescence readers for monitoring caspase activity). Second, virtually all cell biologists are familiar with the observation of cell cultures under the microscope before any sort of experimental intervention, a routine practice that has certainly contributed to the persistence of morphological classifications. Third, it has been assumed for a long time that some degree of morphological uniformity would represent the activation of identical or at least similar lethal signaling cascades. Only recently has it become clear that apparently similar cell death morphotypes most often hide a great degree of functional, biochemical and immunological heterogeneity.5, 8, 14 Moreover, it should always be remembered that the presence of specific morphological features is not sufficient to establish a causal link between a given process and cellular demise.
Biochemical methods for assessing cell death have many advantages over morphological techniques in that they are quantitative, and hence less prone to operator-dependent misinterpretations. However, these methods also have major drawbacks and must be interpreted with caution, especially when single parameters are being investigated.13, 15 Thus, it should always be kept in mind that single biochemical readouts cannot be used as unequivocal indicators of a precise death modality, for a variety of reasons. First, a cell death pathway that is often associated with a particular biochemical process may be normally executed in the absence of this process. Thus, at least in vitro, caspase activation is not a strict requirement for multiple cases of apoptosis (see below).16, 17 Similarly, phosphatidylserine (PS) exposure, which is widely considered as an early marker of apoptotic cell death,18 reportedly does not occur in autophagy-deficient cells succumbing to apoptosis.19 Second, a ‘specific’ cell death-related phenomenon may occur along with the execution of another cell death mode. For instance, excessive generation of reactive oxygen species (ROS) and reactive nitrogen species has been associated with several cases of apoptosis,20, 21, 22 yet it also occurs during regulated necrosis.23, 24 Along similar lines, PS exposure is not a prerogative of apoptotic cell death, as it also constitutes an early feature of parthanatos and netosis (see below).25, 26 Third, a cell death-associated biochemical process can develop at a sublethal or transient level, which does not lead to the cell demise. Thus, while full-blown mitochondrial outer membrane permeabilization (MOMP) constitutes a point-of-no-return of intrinsic apoptosis (see below),20 limited extents of MOMP (i.e., concerning a fraction of the mitochondrial pool) and the consequent (localized) activation of caspase-3 have been shown to participate in several cell death-unrelated programs such as the differentiation of megakaryocytes and granulocytes.27
Definition of ‘Extrinsic Apoptosis’
The term ‘extrinsic apoptosis’ has been extensively used to indicate instances of apoptotic cell death that are induced by extracellular stress signals that are sensed and propagated by specific transmembrane receptors.28, 29, 30 Extrinsic apoptosis can be initiated by the binding of lethal ligands, such as FAS/CD95 ligand (FASL/CD95L), tumor necrosis factor α (TNF_α_) and TNF (ligand) superfamily, member 10 (TNFSF10, best known as TNF-related apoptosis inducing ligand, TRAIL), to various death receptors (i.e., FAS/CD95, TNF_α_ receptor 1 (TNFR1) and TRAIL receptor (TRAILR)1–2, respectively).28 Alternatively, an extrinsic pro-apoptotic signal can be dispatched by the so-called ‘dependence receptors’, including netrin receptors (e.g., UNC5A-D and deleted in colorectal carcinoma, DCC), which only exert lethal functions when the concentration of their specific ligands falls below a critical threshold level.30
One prototypic signaling pathway leading to extrinsic apoptosis is elicited by FAS ligation. In the absence of FASL, FAS subunits spontaneously assemble at the plasma membrane to generate trimers, owing to the so-called pre-ligand assembly domain (PLAD).31 Ligand binding stabilizes such trimers while inducing a conformational change that allows for the assembly of a dynamic multiprotein complex at the cytosolic tail of the receptor. This occurs owing to a conserved sequence of 80 residues that is shared by all death receptors, the so-called ‘death domain’ (DD).32, 33 Proteins recruited at the DD of FAS include receptor-interacting protein kinase 1 (RIPK1, best known as RIP1); FAS-associated protein with a DD (FADD); multiple isoforms of c-FLIP;34, 35 cellular inhibitor of apoptosis proteins (cIAPs), E3 ubiquitin ligases that also inhibit apoptosis owing to their ability to interfere with caspase activation;36 and pro-caspase-8 (or -10).37, 38, 39, 40, 41 The resulting supramolecular complex, which has been dubbed ‘death-inducing signaling complex’ (DISC), constitutes a platform that regulates the activation of caspase-8 (or -10).38, 42
Of note, TNFR1-like proteins also require TNFR-associated DD (TRADD) for recruiting FADD and caspase-8, whereas FAS and TRAILR1/2 do not,29 pointing to the existence of subgroups of death receptors with specific signaling properties. Similarly, the DDs of some death receptors, for instance, TNFR1, recruit several other proteins that are not found at FADD-assembled DISCs, including TNFR-associated factor 2 (TRAF2) and TRAF5.43 In this specific context, RIP1 is polyubiquitinated by cIAPs,44 allowing for the recruitment of transforming growth factor β (TGF_β_)-activated kinase 1 (TAK1), TAK1-binding protein 2 (TAB2) and TAB3, which together can stimulate the canonical activation pathway for the multifunctional transcription factor NF-_κ_B.45 Thus, death receptor activation not always entails the transduction of a lethal signal. This is particularly true for TNFR1, which has been shown to mediate cellular outcomes as different as proliferation and (distinct modalities of) cell death (see below). Irrespective of these variations, both FAS- and TNFR1-elicited signaling pathways appear to be subjected to a consistent degree of regulation upon receptor compartmentalization. A detailed discussion of these aspects goes largely beyond the scope of this paper and can be found in Schutze et al.29
In some cell types including lymphocytes (which have been dubbed ‘type I cells’),46, 47 active caspase-8 directly catalyzes the proteolytic maturation of caspase-3, thereby triggering the executioner phase of caspase-dependent apoptosis in a mitochondrion-independent manner.48 In other cells such as hepatocytes and pancreatic β cells (‘type II cells’),46, 47, 49 caspase-8 mediates the proteolytic cleavage of BH3-interacting domain death agonist (BID), leading to the generation of a mitochondrion-permeabilizing fragment (known as truncated BID, tBID).49, 50, 51 Thus, while type I cells undergo extrinsic apoptosis irrespective of any contribution by mitochondria (tBID and MOMP can occur in these cells, but they are dispensable for the execution of extrinsic apoptosis), type II cells succumb from the activation of death receptors while showing signs of MOMP, including the dissipation of mitochondrial transmembrane potential (Δ_ψ_m) and the release of toxic proteins that are normally retained within the mitochondrial intermembrane space (IMS).20 Among these, cytochrome c (CYTC) drives – together with the cytoplasmic adaptor protein APAF1 and dATP – the assembly of the apoptosome, another caspase-activating multiprotein complex (see below).52 The actual contribution of caspase-10, a close homolog of caspase-8, to extrinsic apoptosis remains unclear. Thus, whereas several reports indicate that caspase-10 is recruited at the DISC and gets activated in response to death receptor signaling,41, 53 it seems that caspase-10 cannot functionally substitute for caspase-8.53 Moreover, it has recently been suggested that caspase-10 might be required for the lethal signaling cascade ignited by death receptors in the presence of caspase inhibitors (see below).54
The molecular routes by which dependence receptors are connected to the rapid activation of executioner caspases, in particular caspase-3, have only recently begun to emerge. Thus, in the absence of their ligands, some dependence receptors like Patched and DCC appear to interact with the cytoplasmic adaptor protein DRAL to assemble a caspase-9-activating platform.55 Another dependence receptor, UNC5B, responds to the withdrawal of netrin-1 by recruiting a signaling complex that includes protein phosphatase 2A (PP2A) and death-associated protein kinase 1 (DAPK1).56 This multiprotein interaction would lead to the PP2A-mediated dephosphorylation of DAPK, in turn unleashing its multifaceted pro-apoptotic potential.57
As a note, there are several other transmembrane proteins that – at least under selected circumstances – can transduce lethal signals in response to ligand binding, including (although presumably not limited to) CD2,58 CD4,59 TNFRSF8/CD30,60, 61 TNFRSF5/CD40,60 CD45,62 CXCR459 and class I/II MHC molecules.63 Similar to TNFR1, most of these proteins have dual functions: depending on the cellular context and triggering stimulus they can engage either pro-survival or pro-death signals. However, the molecular cascades triggered by these receptors are complex and often poorly elucidated, in particular with regard to their dependency on caspases.
On the basis of these considerations, we propose the following operational definition of extrinsic apoptosis. Extrinsic apoptosis is a caspase-dependent cell death subroutine, and hence can be suppressed (at least theoretically) by pan-caspase chemical inhibitors such as _N_-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) or by the overexpression of viral inhibitors of caspases like cytokine response modifier A (CrmA).64 Extrinsic apoptosis would feature one among three major lethal signaling cascades: (i) death receptor signaling and activation of the caspase-8 (or -10) → caspase-3 cascade; (ii) death receptor signaling and activation of the caspase-8 → tBID → MOMP → caspase-9 → caspase-3 pathway; or (iii) ligand deprivation-induced dependence receptor signaling followed by (direct or MOMP-dependent) activation of the caspase-9 → caspase-3 cascade (Table 1 and Figure 1).
Table 1 Functional classification of regulated cell death modes
Figure 1
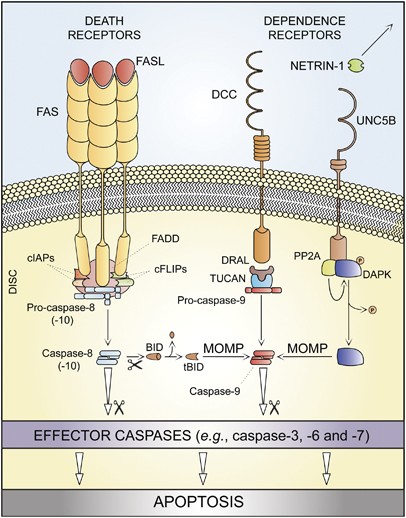
Extrinsic apoptosis. Upon FAS ligand (FASL) binding, the cytoplasmic tails of FAS (also known as CD95, a prototypic death receptor) trimers recruit (among other proteins) FAS-associated protein with a death domain (FADD), cellular inhibitor of apoptosis proteins (cIAPs), c-FLIPs and pro-caspase-8 (or -10). This supramolecular platform, which has been dubbed ‘death-inducing signaling complex’ (DISC), controls the activation of caspase-8 (-10). Within the DISC, c-FLIPs and cIAPs exert pro-survival functions. However, when lethal signals prevail, caspase-8 gets activated and can directly trigger the caspase cascade by mediating the proteolytic maturation of caspase-3 (in type I cells) or stimulate mitochondrial outer membrane permeabilization (MOMP) by cleaving the BH3-only protein BID (in type II cells). Extrinsic apoptosis can also be ignited by dependence receptors like DCC or UNC5B, which relay lethal signals in the absence of their ligand (netrin-1). In the case of DCC and UNC5B, the pro-apoptotic signaling proceeds through the assembly of a DRAL- and TUCAN- (or NLRP1-) containing caspase-9-activating platform or by the dephosphorylation-mediated activation of death-associated protein kinase 1 (DAPK1) by UNC5B-bound protein phosphatase 2A (PP2A), respectively. DAPK1 can mediate the direct activation of executioner caspases or favor MOMP. tBID, truncated BID
Definition of caspase-dependent and caspase-independent ‘intrinsic apoptosis’
The apoptotic demise of cells can be triggered by a plethora of intracellular stress conditions, including DNA damage, oxidative stress, cytosolic Ca2+ overload, mild excitotoxicity (related to glutamate receptor overstimulation in the nervous system), accumulation of unfolded proteins in the endoplasmic reticulum (ER) and many others. Although the signaling cascades that trigger intrinsic apoptosis are highly heterogeneous as far as the initiating stimuli are concerned, they are all wired to a mitochondrion-centered control mechanism.20 Frequently, along with the propagation of the pro-apoptotic signaling cascade, anti-apoptotic mechanisms are also engaged, in an attempt to allow cells to cope with stress. In this scenario, both pro- and anti-apoptotic signals converge at mitochondrial membranes, which become permeabilized when the former predominate over the latter.20 MOMP can start at the outer mitochondrial membrane owing to the pore-forming activity of pro-apoptotic members of the BCL-2 protein family such as BAK and BAX or can result from a phenomenon (the so-called mitochondrial permeability transition, MPT) that originates at the inner mitochondrial membrane due to the opening of a multiprotein complex known as the permeability transition pore complex (PTPC).65, 66 Irrespective of the precise biochemical and physical mechanisms through which it develops, irreversible MOMP affecting most mitochondria within a single cell has multiple lethal consequences: (i) the dissipation of the Δ_ψ_m, with cessation of mitochondrial ATP synthesis and Δ_ψ_m-dependent transport activities; (ii) the release of toxic proteins from the IMS into the cytosol, as this applies to CYTC, apoptosis-inducing factor (AIF), endonuclease G (ENDOG), direct IAP-binding protein with low pI (DIABLO, also known as second mitochondria-derived activator of caspases, SMAC) and high temperature requirement protein A2 (HTRA2); and (iii) the inhibition of the respiratory chain (favored by the loss of CYTC), eliciting or aggravating ROS overproduction and hence activating a feed-forward circuit for the amplification of the apoptotic signal.20
Thus, intrinsic apoptosis results from a bioenergetic and metabolic catastrophe coupled to multiple active executioner mechanisms. Upon MOMP, cytosolic CYTC participates with APAF1 and dATP in the formation of the apoptosome, which triggers the caspase-9 → caspase-3 proteolytic cascade.52, 67 AIF and ENDOG relocate to the nucleus, where they mediate large-scale DNA fragmentation independently of caspases.68, 69, 70, 71 SMAC/DIABLO and HTRA2 inhibit the anti-apoptotic function of several members of the IAP family, thereby derepressing caspase activation.72, 73, 74 In addition, HTRA2 exerts caspase-independent pro-apoptotic effects by virtue of its serine protease activity.75, 76 These mechanisms present a considerable degree of redundancy, as demonstrated by the fact that the knockout or genetic inhibition of single IMS proteins not always affects the execution of intrinsic apoptosis.77 Moreover, the relative contribution of these processes to intrinsic apoptosis varies in distinct physiological, pathological and experimental scenarios. Thus, while ENDOG appears to be dispensable for intrinsic apoptosis in mammalian models,77 Nuc1p, the yeast ortholog of ENDOG, has an important role during the apoptotic response of Saccharomyces cerevisiae to chronological aging in non-fermentable carbon sources (which potentiate mitochondrial respiration).71 DRONC, the ortholog of caspase-9 in Drosophila melanogaster, is required for many forms of developmental cell deaths and apoptosis induced by DNA damage in vivo.78 Conversely, caspase activation seems to have a prominent role in a limited number of instances of stress-induced intrinsic apoptosis in vitro, as demonstrated by the fact that – in contrast to extrinsic apoptosis – chemical and/or genetic inhibition of caspases rarely, if ever, confers long-term cytoprotective effects and truly prevents cell death. In this context, caspase inhibition only delays the execution of cell death, which eventually can even exhibit morphological features of necrosis.16, 17, 79
In view of these observations, we suggest to define ‘intrinsic apoptosis’ as a cell death process that is mediated by MOMP and hence is always associated with (i) generalized and irreversible Δ_ψ_m dissipation, (ii) release of IMS proteins into the cytosol (and their possible relocalization to other subcellular compartments) and (iii) respiratory chain inhibition. We propose to differentiate between caspase-dependent and caspase-independent intrinsic apoptosis based on the extent of cytoprotection conferred by (pharmacological or genetic) inhibition of caspases (Table 1 and Figure 2). This distinction is particularly relevant in vivo, as in some78, 80 (but not all)81 instances of developmental cell death, caspase inhibition has been shown to provide stable cytoprotection. In vitro, in the long run, caspase-independent mechanisms, be they active (e.g., AIF, ENDOG) or passive (e.g., ATP depletion), tend to prevail over caspase inhibition and to kill cells even in instances of intrinsic apoptosis that would have normally been rapidly executed by the caspase cascade. Thus, in vitro, intrinsic apoptosis appears to entail a caspase-dependent and a caspase-independent phase, whose relative contribution to the execution of cell death might be estimated by the extent of short-term (24–48 h) cytoprotection conferred by caspase inhibitors.
Figure 2
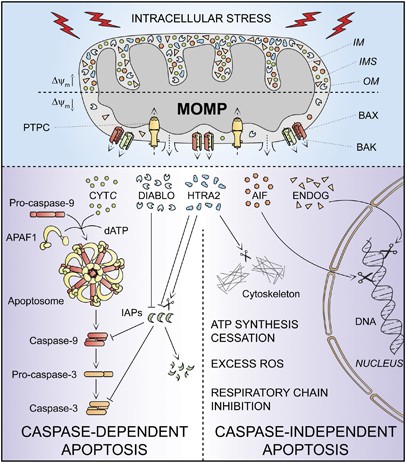
Intrinsic apoptosis. In response to multiple intracellular stress conditions (e.g., DNA damage, cytosolic Ca2+ overload), pro-survival and pro-death signals are generated and converge to a mitochondrion-centered control mechanism. When lethal signals prevail, mitochondrial outer membrane permeabilization (MOMP) occurs and leads to mitochondrial transmembrane potential (Δ_ψ_m) dissipation, arrest of mitochondrial ATP synthesis and Δ_ψ_m-dependent transport activities. Moreover, the respiratory chains gets uncoupled, leading to reactive oxygen species (ROS) overgeneration, and proteins that are normally confined within the mitochondrial intermembrane space (IMS) are released into the cytosol. Among these, cytochrome c (CYTC) drives – together with the cytoplasmic adaptor protein APAF1 and dATP – the assembly of the so-called apoptosome, a multiprotein complex that triggers the caspase-9 → caspase-3 proteolytic cascade. Direct IAP-binding protein with low pI (DIABLO, also known as second mitochondria-derived activator of caspases, SMAC) and high temperature requirement protein A2 (HTRA2) facilitate caspase activation by sequestering and/or degrading several members of the inhibitor of apoptosis protein (IAP) family. On the contrary, apoptosis-inducing factor (AIF) and endonuclease G (ENDOG) function in a caspase-independent manner by relocating to the nucleus and mediating large-scale DNA fragmentation. Of note, the serine protease HTRA2 also contributes to caspase-independent apoptosis by cleaving a wide array of cellular substrates (including cytoskeletal proteins). IM, mitochondrial inner membrane; OM, mitochondrial outer membrane; PTPC, permeability transition pore complex
Definition of ‘Regulated Necrosis’
For a long time, necrosis has been considered as a merely accidental cell death mechanism and was defined by the absence of morphological traits of apoptosis or autophagy.8, 82 Owing to the work of several laboratories,9, 10, 11, 12, 83 it is now clear that necrosis can occur in a regulated manner, and that necrotic cell death has a prominent role in multiple physiological and pathological settings.23 Several triggers can induce regulated necrosis, including alkylating DNA damage, excitotoxins and the ligation of death receptors, at least under selected circumstances.9, 11, 12, 84, 85 Indeed, when caspases (and in particular caspase-8) are inhibited by genetic manipulations (e.g., by gene knockout or RNA interference, RNAi) or blocked by pharmacological agents (e.g., chemical caspase inhibitors), RIP1 and its homolog RIP3 are not degraded and rather engage in physical and functional interactions that ultimately activate the execution of necrotic cell death.9, 11, 12, 23
Regulated necrosis can be further characterized with regard to its dependence on specific signaling modules, and should be named consequently. For instance, cases of regulated necrosis that exhibit RIP1 activation (which can be measured by enzymatic assays or by monitoring RIP1 phosphorylation on S161)11, 86 and that can be suppressed by RIP1 inhibitors including necrostatin-110, 86, 87 should be labeled ‘RIP1-dependent regulated necrosis’. Of note, RIP3-dependent, but RIP1-independent instances of regulated necrosis have recently been identified,12, 88 and these are insensitive to necrostatins.
The term ‘necroptosis’ has recently been used as a synonym of regulated necrosis, but it was originally introduced to indicate a specific case or regulated necrosis, which is ignited by TNFR1 ligation and can be inhibited by the RIP1-targeting chemical necrostatin-1 (Table 1 and Figure 3).10 The NCCD encourages scientists and authors of scientific publications to prefer the use of general versus specific nomenclature (see below). However, ‘necroptosis’ can be used to indicate RIP1- and/or RIP3-dependent regulated necrosis, provided that this expression is explicitly defined at its first appearance and used consistently thereafter.
Figure 3
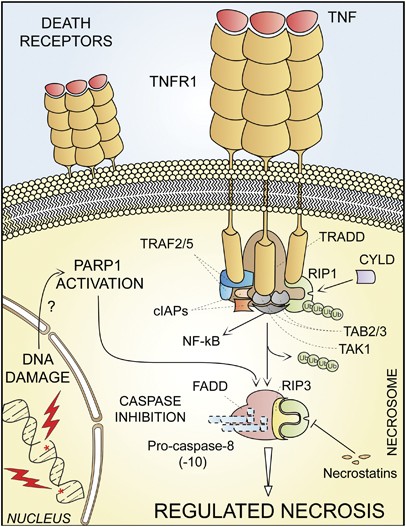
Regulated necrosis. Upon tumor necrosis factor α (TNF_α_) binding, the cytoplasmic tails of TNF receptor 1 (TNFR1, a prototypic death receptor) trimers recruit TNFR-associated death domain (TRADD), receptor-interacting protein kinase 1 (RIP1), cellular inhibitor of apoptosis 1 (cIAP1), cIAP2, TNFR-associated factor 2 (TRAF2) and TRAF5. Within the so-called complex I, RIP1 is polyubiquitinated by cIAPs, thereby providing a docking site for the recruitment of transforming growth factor β (TGF_β_)-activated kinase 1 (TAK1), TAK1-binding protein 2 (TAB2) and TAB3 (which together deliver a pro-survival signal by activating the transcription factor NF-_κ_B). In some pathophysiological and experimental settings, and in particular when caspase-8 is absent or when caspases are inhibited by pharmacological agents, cylindromatosis (CYLD)-deubiquitinated RIP1 engage in physical and functional interactions with its homolog RIP3, ultimately activating the execution of necrotic cell death. Regulated necrosis can also be induced by alkylating DNA damage (possibly by the overactivation of poly(ADP-ribose) polymerase 1, PARP1). In some (but not all) instances, regulated necrosis requires the kinase activity of RIP1, that is, it can be blocked by the RIP1-targeting compounds necrostatins. FADD, FAS-associated protein with a death domain
Definition of ‘Autophagic Cell Death’
On the basis of morphological features, the term ‘autophagic cell death’ has widely been used to indicate instances of cell death that are accompanied by a massive cytoplasmic vacuolization, which often (although not always) indicates increased autophagic flux.5, 8 Although originally this expression did not imply any functional consideration, scientists have quickly adopted the term ‘autophagic cell death’ and used it to imply that autophagy would actually execute the cell demise.89 This applies to at least two very distinct settings. First, autophagy has been shown to mediate physiological cell death in vivo, during the developmental program of D. melanogaster.90, 91, 92 Second, autophagy appears to be responsible for the death of some cancer cells (especially when they lack essential apoptotic modulators like BAX and BAK or caspases)93, 94 that respond to a selected panel of chemotherapeutic agents in vitro.95, 96 Nonetheless, in most known cases, autophagy constitutes a cytoprotective response activated by dying cells in the attempt to cope with stress, and its inhibition accelerates, rather than prevents, cell death.97
Several methods may be used to determine whether the autophagic pathway is activated above baseline levels in the context of the cellular demise. Discussing the advantages and pitfalls of these techniques is beyond the scope of this article, and several excellent reviews on methods for monitoring the autophagic flux have recently been published.15, 98, 99 Nevertheless, it should be remembered that steady-state methods do not provide any reliable estimation of autophagic activity, as they are intrinsically unable to discriminate between enhanced rates of autophagy (increased on-rate) and situations in which the late steps of the pathways are blocked (decreased off-rate).15, 99
From a purely morphological perspective, the term ‘autophagic cell death’ is highly prone to misinterpretation and hence must be used with caution.89 On the contrary, we suggest to reintroduce the term ‘autophagic cell death’ based on biochemical and functional considerations, to indicate a cell death instance that is mediated by autophagy, that is, that can be suppressed by the inhibition of the autophagic pathway by chemicals (e.g., agents that target VPS34) and/or genetic means (e.g., gene knockout/mutation or RNAi targeting essential autophagic modulators like AMBRA1, ATG5, ATG12 or beclin 1100, 101 (Table 1 and Figure 4). As some ATG proteins may have autophagy-independent functions and may even be converted from pro-autophagy to pro-death proteins by proteolytic cleavage (e.g., ATG5 and ATG6),102, 103, 104 it may be advisable to interrogate possible cases of autophagic cell death by knocking down at least two distinct essential autophagic proteins. On the basis of our definition, all cases of cell death that exhibit markers of autophagy such as the lipidation of microtubule-associated protein 1 light chain 3 (better known as LC3/Atg8) or an increased degradation of autophagic substrates like sequestosome 1 (SQSTM1), but cannot be blocked by autophagy inhibition should not be classified as autophagic cell death.
Figure 4
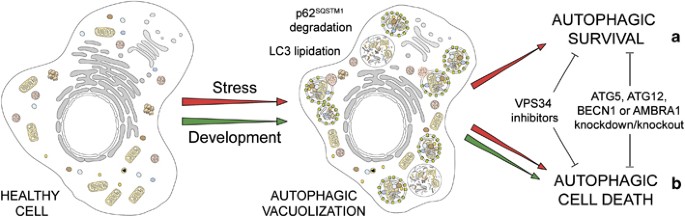
Autophagic cell death. In response to stress and during development, eukaryotic cells often activate autophagy, a mechanism whereby organelles and portion of the cytoplasm are sequestered in double-membraned vesicles (autophagosomes) that are delivered to lysosomes for degradation. Stress-induced autophagy most often exerts cytoprotective functions and favors the re-establishment of homeostasis and survival (a). In this setting, pharmacological or genetic inhibition of autophagy accelerates cell death. On the contrary, these interventions frequently inhibit developmental cell death, indicating that autophagy also constitutes a lethal mechanism that mediates ‘autophagic cell death’ (b)
Definition of ‘Mitotic Catastrophe’
During the past decade, several attempts have been made to delineate the molecular pathways leading to mitotic catastrophe.5, 8, 105, 106 Occasionally, researchers restrictively employ the term ‘mitotic catastrophe’ for cell death occurring in mitosis.106, 107 More frequently, mitotic catastrophe refers to cases of cell death that are triggered by aberrant mitosis and executed either during mitosis or in the subsequent interphase.5, 8 Recently, it has been proposed that mitotic catastrophe might not even constitute a bona fide cell death executioner mechanism, but an oncosuppressive pathway that precedes and is distinct from, yet operates through, cell death or senescence.106, 108
After aberrant mitosis, cells frequently exhibit gross nuclear alterations (e.g., micro- and multinucleation), which have been used as morphological markers for the detection of mitotic catastrophe.8 However, apoptotic and necrotic traits have also been detected in such cells, either concomitant with or following multinucleation.109, 110 Thus, end-point techniques are intrinsically unsuitable for assessing mitotic catastrophe, as they cannot reconstruct the sequence of events that have lead to cell death. To circumvent this issue, novel methods relying on high-throughput video microscopy or time-lapse fluorescence microscopy are under development.99, 111, 112, 113 Several processes were originally associated with and were then shown to be dispensable for (at least some instances of) mitotic catastrophe. These include, but are not limited to, the activation of the DNA damage-responsive protease caspase-2,114 of the tumor suppressor TP53109, 115 and of other members of the TP53 family, including the TP73 variant TAp73.116, 117
In view of recent results from several laboratories indicating that mitotic aberrations can induce cell senescence,118, 119, 120 and that cell death can be either apoptotic or necrotic,8 we have recently proposed a novel definition and categorization of mitotic catastrophe based on purely functional considerations.108 Thus, mitotic catastrophe would not constitute a ‘pure’ cell death executioner pathway, but an oncosuppressive mechanism that: (i) is initiated by perturbations of the mitotic apparatus (i.e., chromosomes and the complex machinery that ensure their faithful segregation); (ii) is initiated during the M phase of the cell cycle; (iii) is paralleled by some degree of mitotic arrest; and (iv) ultimately triggers cell death or senescence (Table 1 and Figure 5). It remains an open conundrum whether the duration of the mitotic arrest truly influences121 or not111 the cell fate after mitotic catastrophe. However, it appears that the crosstalk between TP53- and mammalian target of rapamycin (mTOR)-relayed signals might (at least partially) determine cell senescence.122, 123 In line with our definition, the DNA damage-induced signaling that is initiated at the G2/M transition by the G2 checkpoint (which often, although not always, is mediated by members of the TP53 family)124, 125 does not constitute a case of mitotic catastrophe. Moreover, instances of mitotic arrest that are followed by the re-establishment of homeostasis and resumed proliferation cannot be considered as events of mitotic catastrophe, even when they lead to the gain or loss of chromosomes, and hence to the generation of aneuploid cells.108
Figure 5
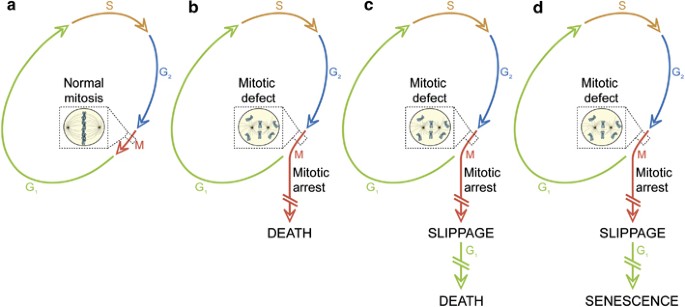
Mitotic catastrophe. (a) In the absence of chemical and genetic perturbations of the mitotic apparatus (including chromosomes and the molecular machinery that ensures their faithful segregation), cells progress through the different phases of the cell cycle to generate a diploid offspring. On the contrary, if chromosomal defects or problems affecting the mitotic machinery are sensed during the M phase, cells become arrested in mitosis due to the activation of mitotic catastrophe (b–d). These cells can undergo different fates: they can die without exiting mitosis (b), reach the G1 phase of the subsequent cell cycle (through a phenomenon that is known as mitotic slippage) and then die (c), or exit mitosis and undergo senescence (d). Irrespective of this diversity of outcomes, mitotic catastrophe can be defined as an oncosuppressive mechanism that precedes and is distinct from, but operates through, cell death and senescence
Tentative Definition of Other Cell Death Modalities
Anoikis
Literally meaning ‘the state of being homeless’, this term of ancient Greek derivation was introduced by Frisch and Francis in 1994 to describe the apoptotic response of adherent cells due to the absence of cell-to-matrix interactions.126 The survival of non-transformed adherent cells does indeed depend on signals transduced by integrins and by some growth factor receptors (such as the epidermal growth factor receptor (EGFR)) upon interaction with the extracellular matrix (ECM).127 As the resistance to anoikis of epithelial cancer cells sustains invasiveness and metastatic potential, great efforts have been undertaken to precisely characterize the underlying molecular cascades.128 On the basis of biochemical and functional considerations, anoikis may currently be defined as an adherent cell-restricted lethal cascade that is ignited by detachment from the matrix and that is characterized by (i) lack of _β_1-integrin engagement, (ii) downregulation of EGFR expression, (iii) inhibition of extracellular-regulated kinase 1 (ERK1) signaling, and (iv) overexpression of the BCL-2 family member BIM (Table 1).127, 129 It should be noted that in most, if not all, instances, the cell death program ignited by anoikis is executed by the molecular machinery for intrinsic apoptosis (see above).127
Entosis
In 2007, Overholtzer et al.130 introduced the term ‘entosis’ to describe a cell death mechanism linked to the ‘cell-in-cell’ phenotype that is frequently exhibited by non-phagocytic cells in clinical tumor samples. Of note, Mormone et al.131 had previously reported a similar phenotype in lymphoblasts from patients with Huntington's disease, and had dubbed it ‘cell cannibalism’.131, 132 Entosis would be provoked by the loss of ECM interaction, but would not entail the activation of apoptotic executioners, thereby constituting a cell death mode distinct from anoikis.130 Recently, the RNAi-mediated downregulation of metallothionein 2A has been shown to promote the ‘cell-in-cell’ phenotype, pointing to a prominent role for adherens junctions in the entotic response.133 Entosis reportedly proceeds in the absence of caspase activation, is insensitive to inhibition by BCL-2 and is paralleled by the activation of the small GTPase RHO and the RHO-associated, coiled-coil containing protein kinase 1 (ROCK1) in the engulfing cell.130, 134 Internalized cells appear virtually normal and later disappear, leading to the hypothesis that they are degraded by lysosomal hydrolases. In some instances, however, such cells have been shown to divide and eventually to be released, raising doubts on the inexorable fate of engulfed cells.130 Conclusive cell fate experiments are urgently awaited to resolve this debate. Meanwhile, we propose to define entosis as an instance of cell death only when all the three following conditions are met. First, the engulfed cells should never exit the phagosome (as detectable by time-lapse microscopy or videomicroscopy) and should be degraded within the lysosome (implying that entosis can be blocked by lysosomal inhibitors). Second, the ‘cell-in-cell’ phenotype should arise from homotypic interactions (i.e., it should involve cells of the same type) and should not engage professional phagocytes. Third, the process should be insensitive to chemical and genetic interventions that normally block caspase-dependent and -independent intrinsic apoptosis (e.g., caspase inhibitors, BCL-2 overexpression) (Table 1).
Parthanatos
Coined after Thanatos, the personification of death in Greek mythology, the term ‘parthanatos’ has been introduced to indicate a particular cell death mode involving the DNA damage-responsive enzymes poly(ADP-ribose) polymerases (PARPs), and in particular PARP1, which alone accounts for more than 90% cellular PARP activity.135 In physiological conditions, PARP1 cooperates with the DNA repair machinery to ensure genomic homeostasis upon mild DNA damage.136 Conversely, PARP1 overactivation has several toxic consequences, including NAD+ and ATP depletion, as well as the accumulation of mitochondriotoxic PAR, which favors Δ_ψ_m dissipation and AIF release.137, 138, 139 Of note, AIF has recently been shown to possess a high-affinity PAR-binding site, and the physical interaction between PAR and AIF appears to be required for parthanatos, both in vitro and in vivo.140 Parthanatos have a role in multiple experimental and physiopathological scenarios, including stroke, diabetes, inflammation and neurodegeneration.141 In line with the original definition of parthanatos, cell death instances should be considered as parthanatic when they depend on early PARP1 activation (i.e., they can be blocked by its chemical and/or genetic inhibition),142 and exhibit NAD+ plus ATP depletion paralleled by AIF-mediated chromatinolysis. Parthanatos constitutes a caspase-independent cell death pathway (Table 1),141 and possibly represents – together with necroptosis – a particular case of regulated necrosis (see above).
Pyroptosis
The term ‘pyroptosis’ has been introduced in 2000 by Brennan and Cookson143 to functionally describe the peculiar death of macrophages infected by Salmonella typhimurium. Several other bacterial triggers of this atypical cell death modality have been identified, including Shigella flexneri, Listeria monocytogenes, Pseudomonas aeruginosa, Francisella tularensis and the Bacillus anthracis toxin.144, 145, 146, 147 However, it has become clear that pyroptosis neither constitutes a macrophage-specific process nor a cell death subroutine that only results from bacterial infection.148 Of note, pyroptotic cells can exhibit apoptotic and/or necrotic morphological features.149
The most distinctive biochemical feature of pyroptosis is the early, induced proximity-mediated activation of caspase-1.150 The pyroptotic activation of caspase-1 can occur in the context of a multiprotein platform known as the inflammasome, which involves the adaptor protein ASC and NOD-like receptors (NLRs) or the cytosolic DNA sensor absent in melanoma 2 (AIM2).147, 151, 152 Alternatively, caspase-1 can be activated by the so-called pyroptosome, a supramolecular assembly of ASC dimers.153 In both cases, active caspase-1 catalyzes the proteolytic maturation and release of pyrogenic interleukin-1_β_ (IL-1_β_) and IL-18.154 Moreover, in some (but not all) instances, this is followed by caspase activation and cell death.143 Active caspase-1 reportedly mediates the proteolytic activation of caspase-7 (rather than that of caspase-3),155 suggesting that pyroptotic cell death might proceed via an unusual caspase-1 → caspase-7 cascade with limited (and perhaps caspase-1-independent) involvement of caspase-3.156 Still, the molecular mechanisms determining why caspase-1 activation sometimes results in cytokine secretion without death and sometimes culminates in the cellular demise remain to be elucidated. We therefore propose to define pyroptosis as a caspase-1-dependent cell death subroutine (i.e., that can be suppressed by the genetic knockout/knockdown of caspase-1 or by caspase-1-specific blockers like N_-benzyloxycarbonyl-Tyr-Val-Ala-DL-Asp-fluoromethylketone (Z-YVAD-fmk)) that is associated with the generation of pyrogenic mediators such as IL-1_β and IL-18 (Table 1). It remains to be clarified whether pyroptosis truly constitutes a cell death subroutine on its own or whether it represents a particular case of caspase-dependent intrinsic apoptosis (see above).
Netosis
In response to several stimuli, neutrophils and eosinophils can release the so-called neutrophil extracellular traps (NETs), that is, microbicidal structures composed of nuclear chromatin, histones and granular antimicrobial proteins.157, 158 Upon the administration of granulocyte–macrophage colony-stimulating factor (GMCSF) or short-term stimulations with the complement fragment C5a, two rather physiological conditions, NETs are generated by viable cells, as demonstrated by several observations: (i) no cytosolic proteins are detected in NETs; (ii) NET-releasing cells fail to take up exclusion dyes; and (iii) NETs can be detected within 30–60 min after stimulation with IL-8 and lipopolysaccharide.158, 159 However, following non-physiological stimulation with phorbol-12-myristate-13-acetate, NETs are released by a subset of neutrophils undergoing a peculiar form of cell death,160 which has been dubbed ‘netosis’.161
Netotic cells exhibit massive vacuolization of the cytoplasm, rapid chromatin decondensation and breakdown of both the nuclear and granular membranes, which is required for proper NET formation.160 Netosis is insensitive to caspase inhibitors and necrostatin-1,162 further demonstrating that it constitutes a cell death subroutine distinct from apoptosis and regulated necrosis. However, the netotic process can be suppressed by pharmacological inhibition of NADPH oxidase (which is responsible for the oxidative burst occurring during neutrophil activation) or autophagy.160, 162 Of note, ROS appear to be required but insufficient for netosis to occur, suggesting that the autophagic component of netosis cannot be entirely attributed to the autophagy-promoting activity of ROS.160, 163 Histone citrullination (i.e., the conversion of positively charged arginine side chains into polar, but uncharged citrulline side chains) also appears to participate in netosis by mediating chromatin decondensation and NET formation.164 Thus, genetic inhibition of the major histone-citrullinating enzyme, that is, peptidylarginine deiminase 4 (PAD4), interfered with NET release by HL-60 cells stimulated with Ca2+ ionophores or Shigella flexneri.164 Importantly, cell death with netotic features has been observed in a subset of cytokine-primed neutrophils following ligation of Siglec-9 and CD44.26, 165 In response to these rather physiological stimuli, dying neutrophils reportedly do not release NETs.26 Thus, whereas NET formation may be paralleled by netosis, this is not always the case: netotic cell death can occur in the absence of NET release and, vice versa, NET can be generated in the absence of cell death.
In view of these observations, netosis might be defined as a cell death subroutine that is: (i) restricted to granulocytic cells; (ii) insensitive to (and perhaps dependent on)163 caspase inhibition; (iii) insensitive to necrostatin; (iv) dependent on NAPDH oxidase-mediated superoxide generation; and (v) dependent on (components of) the autophagic machinery (Table 1). As it stands, netosis shares biochemical features with both autophagic cell death and regulated necrosis. Further investigation is required to elucidate whether netosis is a specific case of either these cell death subroutines or whether it constitutes a cell death mechanism per se.
Cornification
Although cells belonging to the basal layer of the epidermis respond to insults, for instance UV irradiation, by undergoing apoptosis or necrosis, cells of the external layer continuously undergo a physiological cell death subroutine that has been dubbed ‘cornification’.166 This underlies the generation of the stratum corneum, a layer of dead keratinocytes (so-called ‘corneocytes’) containing an mixture of specific proteins (e.g., keratin, loricrin, SPR and involucrin) and lipids (e.g., fatty acids and ceramides) that confers to the skin structural stability, mechanical resistance, elasticity and water repellence.166 Also known as ‘keratinization’ or ‘cornified envelope formation’, cornification is often viewed as a program of terminal differentiation similar to those underlying the maturation of other anucleated tissues (e.g., the lens epithelium, red blood cells).167, 168 All these processes are indeed associated with the (partial) activation of cell death executioners, notably caspases.167, 168, 169 However, lens and red blood cells preserve the ability to succumb from stress-induced cell death,170, 171 whereas corneocytes do not, suggesting that only cornification constitutes a bona fide cell death program.
From a biochemical perspective, cornification is associated with the synthesis of the enzymes and substrates that are required for the generation of the stratum corneum. Enzymes include, although presumably are not limited to, caspase-14172 and transglutaminase (TG)-1, -3 and -5, which catalyze crosslinking reactions.173 Substrates include proteins (e.g., filaggrin, loricrin, SPR, involucrin and SP100), but also lipids, which are extruded into the extracellular space and covalently attached to cornified envelope proteins to ensure skin impermeability.166 The skin of Casp14 −/− mice presents an altered composition of the stratum corneum and is characterized by reduced hydration levels, increased water loss and high sensitivity to UV-induced DNA damage,172 pointing to a critical role for caspase-14 in cornification.
On the basis of these considerations, cornification can be defined as cell death subroutine that: (i) is restricted to keratinocytes; (ii) is functionally linked to the generation of the stratum corneum of the epidermis; and (iii) can be altered, although not blocked, by the inhibition of transglutaminases or caspase-14 (Table 1).
Notes of Caution
There are several issues that should be taken into consideration for appropriately classifying cell death subroutines based on biochemical parameters.
(1) Physiopathological relevance: Here, we provide a functional classification that can be applied to both in vitro and in vivo observations. This said, cell death subroutines should be considered relevant from a physiopathological perspective only when they are shown to occur in vivo and to be critical either for embryonic/post-embryonic development or as an etiological determinant of disease. There probably is a plethora of genes that can influence cell death in physiopathologically relevant settings, but they are never tested under such conditions and most of them remain therefore unidentified. Thus, in vivo studies constitute the ultimate tool to recognize the true importance of cell death signaling pathways and to understand their regulation.
(2) Pharmacological modulators: Chemical agents including the RIP1 inhibitor necrostatin-1, the pan-caspase inhibitor Z-VAD-fmk and the VPS34 inhibitors 3-methyladenine and wortmannin have been widely employed in cell death research, and surely contributed to important discoveries, for example, that of regulated necrosis.10 However, most of these compounds lack adequate specificity to precisely define a cell death program. For instance, Z-VAD-fmk has repeatedly been shown to inhibit non-caspase proteases including calpains.174, 175, 176 Similarly, 3-methyladenine affects multiple facets of cellular metabolism.177 Thus, while tests with pharmacological inhibitors constitute useful starting points for experimentation, they cannot be employed as surrogates of genetic studies based on gene knockout or RNAi.
(3) Specificity of signaling: Most if not all proteins that ignite or mediate cell death subroutines have multiple – and sometimes cell-death-unrelated – functions.27 Thus, whereas cytosolic CYTC activates the apoptosome,52 in the mitochondria CYTC functions as an electron shuttle within the respiratory chain,178 and its complete absence is incompatible with life.179 RIP1 has a plethora of downstream targets that mediate different biological outputs, and not only is it involved in apoptotic and necrotic cell death,23 but is also critical for pro-survival NF-_κ_B signaling.45 Along similar lines, the VPS34 complex regulates autophagy as well as other vesicle trafficking pathways including endocytosis.180 Hence, results coming from either the ablation or genetic inhibition of pleiotropic modulators including (but not limited to) RIP1 and VPS34 should be interpreted with caution, as it is easy to underestimate the number of signaling cascades that have been affected by apparently highly specific perturbations.
(4) Crosstalk between different cell death subroutines: It should always be kept in mind that, in the vast majority of settings (in particular in vivo), cell death subroutines are neither isolated nor mutually exclusive signaling cascades. Most often, pro-survival pathways are engaged along with the propagation of lethal signals. Moreover, stress conditions can result in the activation of multiple lethal mechanisms, which can exhibit variable degrees of overlap. It is the crosstalk between pro-survival and pro-death pathways that determines if and by which subroutine the cell will eventually die. This level of complexity must be taken into account for the classification of cell death modalities, as the inhibition of one specific pathway often unveils the existence of backup mechanisms instead of truly blocking the cell demise.
(5) ‘Programmed’, ‘regulated’ and ‘accidental’ cell death: We suggest to preserve the adjective ‘programmed’ for those physiological instances of cell death – irrespective of the modality by which they are executed – that occur in the context of embryonic/post-embryonic development and tissue homeostasis. ‘Regulated’ should be used to indicate cases of cell death – be they programmed or not – whose initiation and/or execution is mediated by a dedicated molecular machinery, implying that they can be inhibited by targeted pharmacological and/or genetic interventions. Finally, the expression ‘accidental’ should be employed to indicate cell death triggered by extremely harsh physical conditions (e.g., freeze–thawing cycles, high concentrations of pro-oxidants), which cannot be inhibited by pharmacological and/or genetic manipulations and usually exhibits morphological features of necrosis.
(6) General versus specific nomenclature: During the past decade, several neologisms have been introduced to indicate very specific signaling pathways that lead to cell death, including parthanatos, necroptosis, paraptosis, pyronecrosis and several others.5, 6, 8 Although some of these expressions (e.g., necroptosis, parthanatos) have been used in a relatively homogeneous manner, others (e.g., paraptosis, pyronecrosis) have acquired a variety of connotations, facilitating confusion. For this reason, and in line with our mandate (i.e., to provide truly functional definitions of cell death subroutines), we encourage scientists and authors of scientific publications to (i) prefer the use of general terms that bear functional connotations to that of specific names, and (ii) to avoid the introduction of neologisms.
Concluding Remarks
Until now, the field of cell death research has been dominated by morphological definitions that ignore our relentlessly increasing knowledge of the biochemical features of distinct cell death subroutines. Here, the NCCD proposes a new classification of lethal signaling pathways based on biochemical and functional considerations. In this context, ‘loss-of-function’ and ‘gain-of function’ genetic interventions (e.g., RNAi, knockout models and plasmid-driven overexpression systems), as well as chemical inhibitors or activators of important signaling nodes, constitute irreplaceable tools to characterize cell death. During the process of functional characterization, great attention should be paid to ensure that genetic and chemical interventions truly modify the incidence of cell death (as assessed by clonogenic cell survival), rather than activate alternative lethal pathways. With these recommendations in mind and the appropriate tools at hand, researchers have the possibility to label cell death instances with functional and biochemical tags. Our belief is that this classification, if properly applied, will facilitate the understanding of scientific reports, stimulate the communication among scientists and ultimately accelerate the pace of cell death discovery.
Abbreviations
AIM2:
absent in melanoma 2
AIF:
apoptosis-inducing factor
BID:
BH3-interacting domain death agonist
cIAP:
cellular inhibitor of apoptosis protein
CrmA:
cytokine response modifier A
CYTC:
cytochrome c
Δ_ψ_m:
mitochondrial transmembrane potential
DAPK1:
death-associated protein kinase 1
DCC:
deleted in colorectal carcinoma
DD:
death domain
DIABLO:
direct IAP-binding protein with low pI
DISC:
death-inducing signaling complex
EGFR:
epidermal growth factor receptor
ENDOG:
endonuclease G
FADD:
FAS-associated protein with a death domain
GMCSF:
granulocyte-macrophage colony-stimulating factor
HTRA2:
high temperature requirement protein A2
IL:
interleukin
MOMP:
mitochondrial outer membrane permeabilization
mTOR:
mammalian target of rapamycin
NCCD:
Nomenclature Committee on Cell Death
PAR:
poly(ADP-ribose)
NETs:
neutrophil extracellular traps
PARP:
PAR polymerase
PP2A:
protein phosphatase 2A
PS:
phosphatidylserine
RIP:
receptor-interacting protein kinase
RNAi:
RNA interference
ROS:
reactive oxygen species
SMAC:
second mitochondria-derived activator of caspases
ROCK1:
RHO-associated, coiled-coil containing protein kinase 1
SQSTM1:
sequestosome 1
TAB:
TAK1-binding protein
TAK1:
TGF_β_-activated kinase 1
tBID:
truncated BID
TG:
transglutaminase
TGF_β_:
transforming growth factor β
TNF_α_:
tumor necrosis factor α
TNFR:
TNF_α_ receptor
TRADD:
TNFR-associated DD
TRAF:
TNFR-associated factor
TRAIL:
TNF_α_-related apoptosis-inducing ligand
TRAILR:
TRAIL receptor
Z-VAD-fmk:
_N_-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
Z-YVAD-fmk:
_N_-benzyloxycarbonyl-Tyr-Val-Ala-DL-Asp-fluoromethylketone
References
- Lockshin RA, Williams CM . Programmed cell death – II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 1964; 10: 643–649.
CAS Google Scholar - Kerr JF . A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol 1965; 90: 419–435.
CAS PubMed Google Scholar - Lockshin RA, Williams CM . Programmed cell death – I. Cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J Insect Physiol 1965; 11: 123–133.
CAS PubMed Google Scholar - Schweichel JU, Merker HJ . The morphology of various types of cell death in prenatal tissues. Teratology 1973; 7: 253–266.
CAS PubMed Google Scholar - Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14: 1237–1243.
CAS PubMed Google Scholar - Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 2005; 12 (Suppl 2): 1463–1467.
CAS PubMed Google Scholar - Melino G . The Sirens’ song. Nature 2001; 412: 23.
CAS PubMed Google Scholar - Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11.
CAS PubMed Google Scholar - Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
CAS PubMed PubMed Central Google Scholar - Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
CAS PubMed Google Scholar - He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
CAS PubMed Google Scholar - Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
CAS PubMed Google Scholar - Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
CAS PubMed Google Scholar - Green DR, Ferguson T, Zitvogel L, Kroemer G . Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363.
CAS PubMed PubMed Central Google Scholar - Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–175.
CAS PubMed Google Scholar - Chipuk JE, Green DR . Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol 2005; 6: 268–275.
CAS PubMed Google Scholar - Kroemer G, Martin SJ . Caspase-independent cell death. Nat Med 2005; 11: 725–730.
PubMed Google Scholar - Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med 1995; 182: 1545–1556.
CAS PubMed Google Scholar - Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931–946.
CAS PubMed Google Scholar - Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
CAS PubMed Google Scholar - Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA . Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with _N_-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 1995; 92: 7162–7166.
CAS PubMed PubMed Central Google Scholar - Nicotera P, Bernassola F, Melino G . Nitric oxide (NO), a signaling molecule with a killer soul. Cell Death Differ 1999; 6: 931–933.
CAS PubMed Google Scholar - Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
CAS PubMed Google Scholar - Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, Finazzi-Agro A . S-nitrosylation regulates apoptosis. Nature 1997; 388: 432–433.
CAS PubMed Google Scholar - Wang Y, Dawson VL, Dawson TM . Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol 2009; 218: 193–202.
CAS PubMed PubMed Central Google Scholar - Mihalache CC, Yousefi S, Conus S, Villiger PM, Schneider EM, Simon HU . Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J Immunol 2011; 186: 6532–6542.
CAS PubMed Google Scholar - Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM et al. No death without life: vital functions of apoptotic effectors. Cell Death Differ 2008; 15: 1113–1123.
CAS PubMed Google Scholar - Wajant H . The Fas signaling pathway: more than a paradigm. Science 2002; 296: 1635–1636.
CAS PubMed Google Scholar - Schutze S, Tchikov V, Schneider-Brachert W . Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat Rev Mol Cell Biol 2008; 9: 655–662.
PubMed Google Scholar - Mehlen P, Bredesen DE . Dependence receptors: from basic research to drug development. Sci Signal 2011; 4: mr2.
PubMed Google Scholar - Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000; 288: 2354–2357.
CAS PubMed Google Scholar - Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH et al. Self-association of the ‘death domains’ of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J Biol Chem 1995; 270: 387–391.
CAS PubMed Google Scholar - Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME . Apoptosis signaling by death receptors. Eur J Biochem 1998; 254: 439–459.
CAS PubMed Google Scholar - Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997; 386: 517–521.
CAS PubMed Google Scholar - Budd RC, Yeh WC, Tschopp J . cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol 2006; 6: 196–204.
CAS PubMed Google Scholar - Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 1998; 17: 2215–2223.
CAS PubMed PubMed Central Google Scholar - Boldin MP, Goncharov TM, Goltsev YV, Wallach D . Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996; 85: 803–815.
CAS PubMed Google Scholar - Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death – inducing signaling complex. Cell 1996; 85: 817–827.
CAS PubMed Google Scholar - Meier P, Vousden KH . Lucifer's labyrinth – ten years of path finding in cell death. Mol Cell 2007; 28: 746–754.
CAS PubMed Google Scholar - Lavrik I, Golks A, Krammer PH . Death receptor signaling. J Cell Sci 2005; 118: 265–267.
CAS PubMed Google Scholar - Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ . Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci USA 2001; 98: 13884–13888.
CAS PubMed PubMed Central Google Scholar - Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 1995; 14: 5579–5588.
CAS PubMed PubMed Central Google Scholar - Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
CAS PubMed Google Scholar - Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
CAS PubMed Google Scholar - Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ . Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22: 245–257.
CAS PubMed Google Scholar - Barnhart BC, Alappat EC, Peter ME . The CD95 type I/type II model. Semin Immunol 2003; 15: 185–193.
CAS PubMed Google Scholar - Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675–1687.
CAS PubMed PubMed Central Google Scholar - Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES . Molecular ordering of the Fas-apoptotic pathway: the Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA 1996; 93: 14486–14491.
CAS PubMed PubMed Central Google Scholar - Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999; 400: 886–891.
CAS PubMed Google Scholar - Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
CAS PubMed Google Scholar - Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
CAS PubMed Google Scholar - Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–489.
CAS PubMed Google Scholar - Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H . Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J 2002; 21: 4520–4530.
CAS PubMed PubMed Central Google Scholar - Lafont E, Milhas D, Teissie J, Therville N, Andrieu-Abadie N, Levade T et al. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PLoS One 2010; 5: e13638.
PubMed PubMed Central Google Scholar - Mille F, Thibert C, Fombonne J, Rama N, Guix C, Hayashi H et al. The Patched dependence receptor triggers apoptosis through a DRAL–caspase-9 complex. Nat Cell Biol 2009; 11: 739–746.
CAS PubMed PubMed Central Google Scholar - Guenebeaud C, Goldschneider D, Castets M, Guix C, Chazot G, Delloye-Bourgeois C et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell 2010; 40: 863–876.
CAS PubMed Google Scholar - Bialik S, Kimchi A . The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem 2006; 75: 189–210.
CAS PubMed Google Scholar - Wesselborg S, Prufer U, Wild M, Schraven B, Meuer SC, Kabelitz D . Triggering via the alternative CD2 pathway induces apoptosis in activated human T lymphocytes. Eur J Immunol 1993; 23: 2707–2710.
CAS PubMed Google Scholar - Berndt C, Mopps B, Angermuller S, Gierschik P, Krammer PH . CXCR4 and CD4 mediate a rapid CD95-independent cell death in CD4(+) T cells. Proc Natl Acad Sci USA 1998; 95: 12556–12561.
CAS PubMed PubMed Central Google Scholar - Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J 1999; 18: 3034–3043.
CAS PubMed PubMed Central Google Scholar - Krysov SV, Rowley TF, Al-Shamkhani A . Inhibition of p38 mitogen-activated protein kinase unmasks a CD30-triggered apoptotic pathway in anaplastic large cell lymphoma cells. Mol Cancer Ther 2007; 6: 703–711.
CAS PubMed Google Scholar - Lesage S, Steff AM, Philippoussis F, Page M, Trop S, Mateo V et al. CD4+ CD8+ thymocytes are preferentially induced to die following CD45 cross-linking, through a novel apoptotic pathway. J Immunol 1997; 159: 4762–4771.
CAS PubMed Google Scholar - Woodle ES, Smith DM, Bluestone JA, Kirkman III WM, Green DR, Skowronski EW . Anti-human class I MHC antibodies induce apoptosis by a pathway that is distinct from the Fas antigen-mediated pathway. J Immunol 1997; 158: 2156–2164.
CAS PubMed Google Scholar - Gagliardini V, Fernandez PA, Lee RK, Drexler HC, Rotello RJ, Fishman MC et al. Prevention of vertebrate neuronal death by the crmA gene. Science 1994; 263: 826–828.
CAS PubMed Google Scholar - Tait SW, Green DR . Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11: 621–632.
CAS PubMed Google Scholar - Brenner C, Grimm S . The permeability transition pore complex in cancer cell death. Oncogene 2006; 25: 4744–4756.
CAS PubMed Google Scholar - Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome _c_-dependent activation of caspase-3. Cell 1997; 90: 405–413.
CAS PubMed Google Scholar - Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001; 410: 549–554.
CAS PubMed Google Scholar - Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999; 397: 441–446.
CAS PubMed Google Scholar - Li LY, Luo X, Wang X . Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001; 412: 95–99.
CAS PubMed Google Scholar - Buttner S, Eisenberg T, Carmona-Gutierrez D, Ruli D, Knauer H, Ruckenstuhl C et al. Endonuclease G regulates budding yeast life and death. Mol Cell 2007; 25: 233–246.
PubMed Google Scholar - Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y . Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000; 406: 855–862.
CAS PubMed Google Scholar - Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C . Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev 2003; 17: 1487–1496.
CAS PubMed PubMed Central Google Scholar - Srinivasula SM, Gupta S, Datta P, Zhang Z, Hegde R, Cheong N et al. Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2. J Biol Chem 2003; 278: 31469–31472.
CAS PubMed Google Scholar - Vande Walle L, Van Damme P, Lamkanfi M, Saelens X, Vandekerckhove J, Gevaert K et al. Proteome-wide identification of HtrA2/Omi substrates. J Proteome Res 2007; 6: 1006–1015.
CAS PubMed Google Scholar - Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein–caspase interaction. J Biol Chem 2002; 277: 432–438.
CAS PubMed Google Scholar - David KK, Sasaki M, Yu SW, Dawson TM, Dawson VL . EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ 2006; 13: 1147–1155.
CAS PubMed Google Scholar - Daish TJ, Mills K, Kumar S . Drosophila caspase DRONC is required for specific developmental cell death pathways and stress-induced apoptosis. Dev Cell 2004; 7: 909–915.
CAS PubMed Google Scholar - Lemaire C, Andreau K, Souvannavong V, Adam A . Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett 1998; 425: 266–270.
CAS PubMed Google Scholar - Yuan JY, Horvitz HR . The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol 1990; 138: 33–41.
CAS PubMed Google Scholar - Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P . Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol 1999; 9: 967–970.
CAS PubMed Google Scholar - Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G . Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis 2007; 12: 803–813.
CAS PubMed Google Scholar - Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135: 1311–1323.
CAS PubMed PubMed Central Google Scholar - Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB . Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004; 18: 1272–1282.
CAS PubMed PubMed Central Google Scholar - Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005; 120: 275–285.
CAS PubMed Google Scholar - Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313–321.
CAS PubMed PubMed Central Google Scholar - Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C . Structure–activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg Med Chem Lett 2007; 17: 1455–1465.
CAS PubMed Google Scholar - Upton JW, Kaiser WJ, Mocarski ES . Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010; 7: 302–313.
CAS PubMed PubMed Central Google Scholar - Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–1010.
CAS PubMed PubMed Central Google Scholar - Berry DL, Baehrecke EH . Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131: 1137–1148.
CAS PubMed PubMed Central Google Scholar - Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol 2009; 19: 1741–1746.
CAS PubMed PubMed Central Google Scholar - Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol 2010; 190: 523–531.
CAS PubMed PubMed Central Google Scholar - Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6: 1221–1228.
CAS PubMed Google Scholar - Fazi B, Bursch W, Fimia GM, Nardacci R, Piacentini M, Di Sano F et al. Fenretinide induces autophagic cell death in caspase-defective breast cancer cells. Autophagy 2008; 4: 435–441.
CAS PubMed Google Scholar - Grander D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T . Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. Autophagy 2009; 5: 1198–1200.
CAS PubMed Google Scholar - Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ 2009; 16: 1018–1029.
CAS PubMed Google Scholar - Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
CAS PubMed PubMed Central Google Scholar - Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
CAS PubMed PubMed Central Google Scholar - Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G . Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10: 221–237.
CAS PubMed Google Scholar - Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447: 1121–1125.
CAS PubMed Google Scholar - Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–676.
CAS PubMed Google Scholar - Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA, Kim JC . Caspase-mediated cleavage of ATG6/beclin-1 links apoptosis to autophagy in HeLa cells. Cancer Lett 2009; 274: 95–100.
CAS PubMed Google Scholar - Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124–1132.
CAS PubMed Google Scholar - Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I et al. Caspase-mediated cleavage of beclin-1 inactivates beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010; 1: e18.
CAS PubMed PubMed Central Google Scholar - Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G . Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–2837.
CAS PubMed Google Scholar - Vakifahmetoglu H, Olsson M, Zhivotovsky B . Death through a tragedy: mitotic catastrophe. Cell Death Differ 2008; 15: 1153–1162.
CAS PubMed Google Scholar - Roninson IB, Broude EV, Chang BD . If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4: 303–313.
CAS PubMed Google Scholar - Vitale I, Galluzzi L, Castedo M, Kroemer G . Mitotic catastrophe: a mechanism for avoiding of genomic instability. Nat Rev Mol Cell Biol 2011; 12: 385–392.
CAS PubMed Google Scholar - Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P et al. Apoptosis regulation in tetraploid cancer cells. EMBO J 2006; 25: 2584–2595.
CAS PubMed PubMed Central Google Scholar - Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B . DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15: 555–566.
CAS PubMed Google Scholar - Gascoigne KE, Taylor SS . Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14: 111–122.
CAS PubMed Google Scholar - Rello-Varona S, Kepp O, Vitale I, Michaud M, Senovilla L, Jemaa M et al. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis 2010; 1: e25.
CAS PubMed PubMed Central Google Scholar - Imreh G, Vakifahmetoglu Norberg H, Imreh S, Zhivotovsky B . Chromosomal breaks during mitotic catastrophe trigger _γ_H2AX–ATM–p53-mediated apoptosis. J Cell Sci 2011 (in press).
- Vakifahmetoglu-Norberg H, Zhivotovsky B . The unpredictable caspase-2: what can it do? Trends Cell Biol 2010; 20: 150–159.
CAS PubMed Google Scholar - Sedic M, Poznic M, Gehrig P, Scott M, Schlapbach R, Hranjec M et al. Differential antiproliferative mechanisms of novel derivative of benzimidazo[1,2-alpha]quinoline in colon cancer cells depending on their p53 status. Mol Cancer Ther 2008; 7: 2121–2132.
CAS PubMed Google Scholar - Tomasini R, Tsuchihara K, Tsuda C, Lau SK, Wilhelm M, Ruffini A et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci USA 2009; 106: 797–802.
CAS PubMed PubMed Central Google Scholar - Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22: 2677–2691.
CAS PubMed PubMed Central Google Scholar - Eom YW, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S et al. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005; 24: 4765–4777.
CAS PubMed Google Scholar - Gao RJ, Liang YX, Li DD, Zhang HY, Zhen YS . Effect of lidamycin on telomerase activity in human hepatoma BEL-7402 cells. Biomed Environ Sci 2007; 20: 189–197.
CAS PubMed Google Scholar - Yun M, Han YH, Yoon SH, Kim HY, Kim BY, Ju YJ et al. p31comet induces cellular senescence through p21 accumulation and Mad2 disruption. Mol Cancer Res 2009; 7: 371–382.
CAS PubMed Google Scholar - Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV . Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene 2008; 27: 4402–4410.
CAS PubMed Google Scholar - Galluzzi L, Kepp O, Kroemer G . TP53 and MTOR crosstalk to regulate cellular senescence. Aging (Albany, NY) 2010; 2: 535–537.
CAS Google Scholar - Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV . The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany, NY) 2010; 2: 344–352.
CAS Google Scholar - Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G . The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12: 259–265.
CAS PubMed Google Scholar - Tomasini R, Mak TW, Melino G . The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol 2008; 18: 244–252.
CAS PubMed Google Scholar - Frisch SM, Francis H . Disruption of epithelial cell–matrix interactions induces apoptosis. J Cell Biol 1994; 124: 619–626.
CAS PubMed Google Scholar - Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol 2003; 5: 733–740.
CAS PubMed Google Scholar - Frisch SM, Screaton RA . Anoikis mechanisms. Curr Opin Cell Biol 2001; 13: 555–562.
CAS PubMed Google Scholar - Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS . BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell 2007; 12: 221–234.
CAS PubMed PubMed Central Google Scholar - Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007; 131: 966–979.
CAS PubMed Google Scholar - Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG et al. Genotype-dependent priming to self- and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with Huntington's disease. J Neurochem 2006; 98: 1090–1099.
CAS PubMed Google Scholar - Matarrese P, Ciarlo L, Tinari A, Piacentini M, Malorni W . Xeno-cannibalism as an exacerbation of self-cannibalism: a possible fruitful survival strategy for cancer cells. Curr Pharm Des 2008; 14: 245–252.
CAS PubMed Google Scholar - Lai Y, Lim D, Tan PH, Leung TK, Yip GW, Bay BH . Silencing the metallothionein-2A gene induces entosis in adherent MCF-7 breast cancer cells. Anat Rec (Hoboken) 2010; 293: 1685–1691.
CAS Google Scholar - Fiorentini C, Falzano L, Fabbri A, Stringaro A, Logozzi M, Travaglione S et al. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol Biol Cell 2001; 12: 2061–2073.
CAS PubMed PubMed Central Google Scholar - Ame JC, Spenlehauer C, de Murcia G . The PARP superfamily. Bioessays 2004; 26: 882–893.
CAS PubMed Google Scholar - Jeggo PA . DNA repair: PARP – another guardian angel? Curr Biol 1998; 8: R49–R51.
CAS PubMed Google Scholar - Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA 2006; 103: 18308–18313.
CAS PubMed PubMed Central Google Scholar - Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 2006; 103: 18314–18319.
CAS PubMed PubMed Central Google Scholar - Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
CAS PubMed Google Scholar - Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent dell death (parthanatos). Sci Signal 2011; 4: ra20.
PubMed PubMed Central Google Scholar - David KK, Andrabi SA, Dawson TM, Dawson VL . Parthanatos, a messenger of death. Front Biosci 2009; 14: 1116–1128.
CAS Google Scholar - Virag L, Szabo C . The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 2002; 54: 375–429.
CAS PubMed Google Scholar - Brennan MA, Cookson BT . Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 2000; 38: 31–40.
CAS PubMed Google Scholar - Fink SL, Bergsbaken T, Cookson BT . Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA 2008; 105: 4312–4317.
CAS PubMed PubMed Central Google Scholar - Fink SL, Cookson BT . Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol 2007; 9: 2562–2570.
CAS PubMed Google Scholar - Cervantes J, Nagata T, Uchijima M, Shibata K, Koide Y . Intracytosolic Listeria monocytogenes induces cell death through caspase-1 activation in murine macrophages. Cell Microbiol 2008; 10: 41–52.
CAS PubMed Google Scholar - Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 2010; 11: 385–393.
CAS PubMed PubMed Central Google Scholar - Bergsbaken T, Fink SL, Cookson BT . Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7: 99–109.
CAS PubMed PubMed Central Google Scholar - Kepp O, Galluzzi L, Zitvogel L, Kroemer G . Pyroptosis – a cell death modality of its kind? Eur J Immunol 2010; 40: 627–630.
CAS PubMed Google Scholar - Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A . The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA 1999; 96: 2396–2401.
CAS PubMed PubMed Central Google Scholar - Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G . The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009; 10: 241–247.
CAS PubMed PubMed Central Google Scholar - Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES . AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513.
CAS PubMed PubMed Central Google Scholar - Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 2007; 14: 1590–1604.
CAS PubMed Google Scholar - Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S et al. Caspase-1 activation of IL-1beta and IL-18 are essential for _Shigella flexneri_-induced inflammation. Immunity 2000; 12: 581–590.
CAS PubMed Google Scholar - Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 2008; 7: 2350–2363.
CAS PubMed PubMed Central Google Scholar - Rintahaka J, Lietzen N, Ohman T, Nyman TA, Matikainen S . Recognition of cytoplasmic RNA results in cathepsin-dependent inflammasome activation and apoptosis in human macrophages. J Immunol 2011; 186: 3085–3092.
CAS PubMed Google Scholar - Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 2008; 14: 949–953.
CAS PubMed Google Scholar - Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535.
CAS PubMed Google Scholar - Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU . Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 2009; 16: 1438–1444.
CAS PubMed Google Scholar - Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176: 231–241.
CAS PubMed PubMed Central Google Scholar - Steinberg BE, Grinstein S . Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE 2007; 2007: pe11.
PubMed Google Scholar - Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2010; 21: 290–304.
PubMed PubMed Central Google Scholar - Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T . Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 2011; 18: 581–588.
CAS PubMed PubMed Central Google Scholar - Wang Y, Li M, Stadler S, Correll S, Li P, Wang D et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009; 184: 205–213.
CAS PubMed PubMed Central Google Scholar - von Gunten S, Yousefi S, Seitz M, Jakob SM, Schaffner T, Seger R et al. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 2005; 106: 1423–1431.
CAS PubMed Google Scholar - Candi E, Schmidt R, Melino G . The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005; 6: 328–340.
CAS PubMed Google Scholar - Counis MF, Chaudun E, Arruti C, Oliver L, Sanwal M, Courtois Y et al. Analysis of nuclear degradation during lens cell differentiation. Cell Death Differ 1998; 5: 251–261.
CAS PubMed Google Scholar - Testa U . Apoptotic mechanisms in the control of erythropoiesis. Leukemia 2004; 18: 1176–1199.
CAS PubMed Google Scholar - Lippens S, Kockx M, Knaapen M, Mortier L, Polakowska R, Verheyen A et al. Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death Differ 2000; 7: 1218–1224.
CAS PubMed Google Scholar - Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, Huber SM et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem 2005; 15: 195–202.
CAS PubMed Google Scholar - Yan Q, Liu JP, Li DW . Apoptosis in lens development and pathology. Differentiation 2006; 74: 195–211.
CAS PubMed Google Scholar - Denecker G, Hoste E, Gilbert B, Hochepied T, Ovaere P, Lippens S et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol 2007; 9: 666–674.
CAS PubMed Google Scholar - Steven AC, Steinert PM . Protein composition of cornified cell envelopes of epidermal keratinocytes. J Cell Sci 1994; 107 (Part 2): 693–700.
CAS PubMed Google Scholar - Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffmann SN et al. Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology 2005; 49: 695–702.
CAS PubMed Google Scholar - Waterhouse NJ, Finucane DM, Green DR, Elce JS, Kumar S, Alnemri ES et al. Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ 1998; 5: 1051–1061.
CAS PubMed Google Scholar - Wolf BB, Goldstein JC, Stennicke HR, Beere H, Amarante-Mendes GP, Salvesen GS et al. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood 1999; 94: 1683–1692.
CAS PubMed Google Scholar - Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ . 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem 1988; 175: 325–329.
CAS PubMed Google Scholar - Rich P . Chemiosmotic coupling: the cost of living. Nature 2003; 421: 583.
CAS PubMed Google Scholar - Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 2000; 101: 389–399.
CAS PubMed Google Scholar - Juhasz G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol 2008; 181: 655–666.
CAS PubMed PubMed Central Google Scholar
Acknowledgements
The NCCD acknowledges the valuable input of all Editors of Cell Death and Differentiation as well as that of numerous colleagues that helped shaping the present recommendations in scientific meetings. LG is financed by APO-SYS. DCR is a Wellcome Trust Senior Fellow.
Author information
Authors and Affiliations
- INSERM U848, ‘Apoptosis, Cancer and Immunity’, Villejuif, 94805, France
L Galluzzi, I Vitale, O Kepp & G Kroemer - Institut Gustave Roussy, Villejuif, 94805, France
L Galluzzi, I Vitale & O Kepp - Université Paris Sud-XI, Villejuif, 94805, France
L Galluzzi, I Vitale & O Kepp - Department of Cell Biology, UT Southwestern Medical Center, Dallas, 75390, TX, USA
J M Abrams - Department of Biochemistry and Molecular Biology, Center for Apoptosis Research, Kimmel Cancer Institute, Thomas Jefferson University, Philadelphia, PA 19107, USA,
E S Alnemri - Department of Cancer Biology, University of Massachusetts Medical School, Worcester, 01605, MA, USA
E H Baehrecke - Department of Cell Stress Biology, Roswell Park Cancer Institute, Buffalo, 14263, NY, USA
M V Blagosklonny - Neuroregeneration and Stem Cell Programs, Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
T M Dawson & V L Dawson - Cancer Institute Penn State, Hershey Medical Center, Philadelphia, 17033, PA, USA
W S El-Deiry - Institute for Experimental Cancer Research in Pediatrics, Goethe University, Frankfurt, 60528, Germany
S Fulda - The Beatson Institute for Cancer Research, Glasgow, G61 1BD, UK
E Gottlieb - Department of Immunology, St. Jude Children's Research Hospital, Memphis, 38105, TN, USA
D R Green - Institute of Molecular Life Sciences, University of Zurich, Zurich, 8057, Switzerland
M O Hengartner - Institute of Child Health, University College London, London, WC1N 3JH, UK
R A Knight - Centre for Cancer Biology, SA Pathology, Adelaide, 5000, South Australia, Australia
S Kumar - Department of Medicine, University of Adelaide, Adelaide, 5005, South Australia, Australia
S Kumar - Sanford-Burnham Medical Research Institute, San Diego, 92037, CA, USA
S A Lipton - Salk Institute for Biological Studies, La Jolla, 92037, CA, USA
S A Lipton - The Scripps Research Institute, La Jolla, 92037, CA, USA
S A Lipton - Univerisity of California, San Diego, La Jolla, 92093, CA, USA
S A Lipton - Ludwig Institute for Cancer Research, Oxford, OX3 7DQ, UK
X Lu - Institute of Molecular Biosciences, University of Graz, Graz, 8010, Austria
F Madeo - Department of Therapeutic Research and Medicines Evaluation, Section of Cell Aging and Degeneration, Istituto Superiore di Sanità, Rome, 00161, Italy
W Malorni - Istituto San Raffaele Sulmona, Sulmona, 67039, Italy
W Malorni - Apoptosis, Cancer and Development, CRCL, Lyon, 69008, France
P Mehlen - INSERM, U1052, Lyon, 69008, France
P Mehlen - CNRS, UMR5286, Lyon, 69008, France
P Mehlen - Centre Léon Bérard, Lyon, 69008, France
P Mehlen - University of Michigan Medical School, Ann Arbor, 48109, MI, USA
G Nuñez - Northwestern University Feinberg School of Medicine, Chicago, 60637, IL, USA
M E Peter - Laboratory of Cell Biology, National Institute for Infectious Diseases IRCCS ‘L Spallanzani’, Rome, 00149, Italy
M Piacentini - Department of Biology, University of Rome ‘Tor Vergata’, Rome, 00133, Italy
M Piacentini - Cambridge Institute for Medical Research, Cambridge, CB2 0XY, UK
D C Rubinsztein - Shanghai Institutes for Biological Sciences, Shanghai, 200031, China
Y Shi - Institute of Pharmacology, University of Bern, Bern, 3010, Switzerland
H-U Simon - Department for Molecular Biology, Gent University, Gent, 9052, Belgium
P Vandenabeele - Department for Molecular Biomedical Research, VIB, 9052 Gent, Belgium,
P Vandenabeele - The Cancer Institute of New Jersey, New Brunswick, 08903, NJ, USA
E White - Department of Cell Biology, Harvard Medical School, Boston, 02115, MA, USA
J Yuan - Division of Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, 17177, Sweden
B Zhivotovsky - Department of Experimental Medicine, Biochemical Laboratory IDI-IRCCS, University of Rome ‘Tor Vergata’, Rome, 00133, Italy
G Melino - Medical Research Council, Toxicology Unit, Leicester University, Leicester, LE1 9HN, UK
G Melino - Metabolomics Platform, Institut Gustave Roussy, Villejuif, 94805, France
G Kroemer - Centre de Recherche des Cordeliers, Paris, 75005, France
G Kroemer - Pôle de Biologie, Hôpital Européen Georges Pompidou, AP-HP, 75908 Paris, France,
G Kroemer - Université Paris Descartes, Paris 5, Paris, 75270, France
G Kroemer
Authors
- L Galluzzi
You can also search for this author inPubMed Google Scholar - I Vitale
You can also search for this author inPubMed Google Scholar - J M Abrams
You can also search for this author inPubMed Google Scholar - E S Alnemri
You can also search for this author inPubMed Google Scholar - E H Baehrecke
You can also search for this author inPubMed Google Scholar - M V Blagosklonny
You can also search for this author inPubMed Google Scholar - T M Dawson
You can also search for this author inPubMed Google Scholar - V L Dawson
You can also search for this author inPubMed Google Scholar - W S El-Deiry
You can also search for this author inPubMed Google Scholar - S Fulda
You can also search for this author inPubMed Google Scholar - E Gottlieb
You can also search for this author inPubMed Google Scholar - D R Green
You can also search for this author inPubMed Google Scholar - M O Hengartner
You can also search for this author inPubMed Google Scholar - O Kepp
You can also search for this author inPubMed Google Scholar - R A Knight
You can also search for this author inPubMed Google Scholar - S Kumar
You can also search for this author inPubMed Google Scholar - S A Lipton
You can also search for this author inPubMed Google Scholar - X Lu
You can also search for this author inPubMed Google Scholar - F Madeo
You can also search for this author inPubMed Google Scholar - W Malorni
You can also search for this author inPubMed Google Scholar - P Mehlen
You can also search for this author inPubMed Google Scholar - G Nuñez
You can also search for this author inPubMed Google Scholar - M E Peter
You can also search for this author inPubMed Google Scholar - M Piacentini
You can also search for this author inPubMed Google Scholar - D C Rubinsztein
You can also search for this author inPubMed Google Scholar - Y Shi
You can also search for this author inPubMed Google Scholar - H-U Simon
You can also search for this author inPubMed Google Scholar - P Vandenabeele
You can also search for this author inPubMed Google Scholar - E White
You can also search for this author inPubMed Google Scholar - J Yuan
You can also search for this author inPubMed Google Scholar - B Zhivotovsky
You can also search for this author inPubMed Google Scholar - G Melino
You can also search for this author inPubMed Google Scholar - G Kroemer
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toG Kroemer.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by V De Laurenzi
Rights and permissions
About this article
Cite this article
Galluzzi, L., Vitale, I., Abrams, J. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012.Cell Death Differ 19, 107–120 (2012). https://doi.org/10.1038/cdd.2011.96
- Received: 16 May 2011
- Accepted: 13 June 2011
- Published: 15 July 2011
- Issue Date: January 2012
- DOI: https://doi.org/10.1038/cdd.2011.96