6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27 (original) (raw)
Main
The homodimeric bifunctional 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB) phosphorylate fructose 6-phosphate (F6P) to fructose-2,6-bisphosphate (F2,6BP), which in turn activates 6-phosphofructo-1-kinase and glycolytic flux to lactate.1 Of the four genes encoding distinct PFKFB isozymes (PFKFB1-4), PFKFB3 is distinguished by the presence of multiple copies of the AUUUA instability motif in its 3’untranslated region,2 a very high kinase:phosphatase activity ratio (740 : 1),3 increased protein expression in rapidly proliferating transformed cells,2 solid tumors and leukemias2, 4, 5 and regulation by several proteins essential for tumor progression (e.g. HIF-1_α_,6 Akt7 and PTEN8, 9). Not surprisingly, heterozygous genomic deletion of the pfkfb3 gene has been found to reduce both the glucose metabolism and growth of Ras-transformed tumors in syngeneic mice.10
In recent studies, we unexpectedly observed that PFKFB3 trafficked to the nucleus of multiple cell lines via a highly conserved nuclear localization motif in the C-terminal domain.11 Although the precise role of nuclear PFKFB3 is unknown, ectopic expression of wild-type PFKFB3 in the nucleus was found to stimulate cellular proliferation without affecting glycolysis, suggesting a novel role for nuclear F2,6BP in regulating the cell cycle.11 Moreover, the addition of F2,6BP to total cell lysates was found to increase the cyclin-dependent kinase (Cdk)-dependent phosphorylation of its substrate p27 at threonine 187 (T187), a posttranslational modification that targets p27 for degradation (i.e. high Cdk activity suppresses p27 levels).11 Given that p27 can potently block the G1/S transition and stimulate apoptosis, these data indicated that PFKFB3-mediated production of F2,6BP in the nucleus may directly stimulate Cdks to phosphorylate T187-p27, targeting p27 for degradation by the proteasome and allowing cells to both proliferate and evade apoptosis. Furthermore, these data signified that PFKFB3 may not only be essential for the regulation of glycolysis in the cytoplasm but also for the control of the cell cycle in the nucleus.
Based on these prior studies, we postulated that selective inhibition of PFKFB3 would suppress Cdk1 activity, which in turn would reduce the phosphorylation of T187-p27, resulting in increased p27 expression, reduced G1/S transition and increased apoptosis. We provide evidence to support this chain of biochemical and cellular events after PFKFB3 inhibition as well as direct verification that p27 itself is required for the simultaneous suppression of G1/S transition and induction of apoptosis caused by PFKFB3 inhibition. Given that PFKFB3 inhibitors are entering phase I trials for the treatment of advanced cancers,12 we believe that this new mechanism of action may facilitate the development of rational phase I/II trials that combine other apoptosis-activating agents that disrupt p27 function (e.g. Cdk1 inhibitors) as well as potential biomarkers such as p27 that may demonstrate the on-target effects of PFKFB3 inhibitors in biopsies and resected tumors. From a broader perspective, these data provide further support for the concept that PFKFB3 may be an essential coupler of glucose metabolism and cell cycle progression.
Results
PFKFB3 inhibition suppresses F2,6BP, glucose uptake and glycolysis
We selected HeLa cells to initially conduct a detailed study of the role of PFKFB3 in regulating Cdk1- and p27-mediated G1/S blockade and apoptosis, as the nuclear localization of PFKFB3 was discovered using these cells as a model system.11 We identified two separate siRNA molecules (P31 and P32) that were effective at suppressing PFKFB3 mRNA and protein expression in HeLa cells (Figures 1a–c). As PFKFB3 has the highest kinase:phosphatase ratio of the PFKFB family members, we expected that selective PFKFB3 inhibition in HeLa cells would drop the steady-state concentration of F2,6BP, which, in turn, would suppress both glucose uptake (through feedback allosteric inhibition of hexokinase by glucose 6-phosphate, which is in equilibrium with the substrate of Pfk-1, F6P, via phosphoglucose isomerase) and glycolysis downstream of Pfk-1. We found that both PFKFB3 siRNA molecules potently reduced F2,6BP (Figure 1d), glucose uptake (Figure 1e) and glycolysis as measured by the release of 3H2O from 5-3H-glucose via enolase (Figure 1f). These data confirm our prior observations that PFKFB3 expression is required to maintain glycolytic flux in transformed cells.10
Figure 1
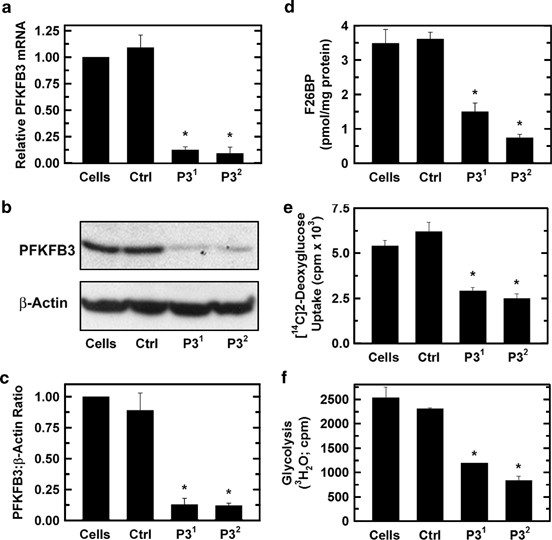
Selective inhibition of PFKFB3 reduces F2,6BP, glucose uptake and glycolysis in HeLa cells. HeLa cells were transfected with a control siRNA (Ctrl) or two PFKFB3 siRNA molecules (P31 and P32) and PFKFB3 mRNA by real-time RT-PCR (a), PFKFB3 protein by western blot (b) and densitometry (c), F2,6BP (d), [14C]-2-deoxy-glucose uptake (e) and [3H]-5-glucose utilization (f) were assessed. *_P-_value<0.05
PFKFB3 inhibition completely suppresses cell proliferation and results in increased early and late apoptotic cells
We next examined the effects of PFKFB3 silencing on exponentially growing HeLa cells by trypan blue exclusion over 24–72 h and observed a complete suppression of cell proliferation after 48 and 72 h (Figure 2a). Interestingly, this antiproliferative effect far exceeded the antiglycolytic effect as detailed in Figures 1e and f. The reduced viable cell number was due to a combination of a G1/S transition block (Figures 2b and d) as has been previously reported after PFKFB3 siRNA transfection13 and by an increase in early and late apoptotic cells (Figures 2c and e) as has been reported after exposure to small molecule inhibitors of PFKFB3.12 These results confirm that PFKFB3 expression is required for both cell cycle progression and maintenance of an antiapoptotic state.
Figure 2
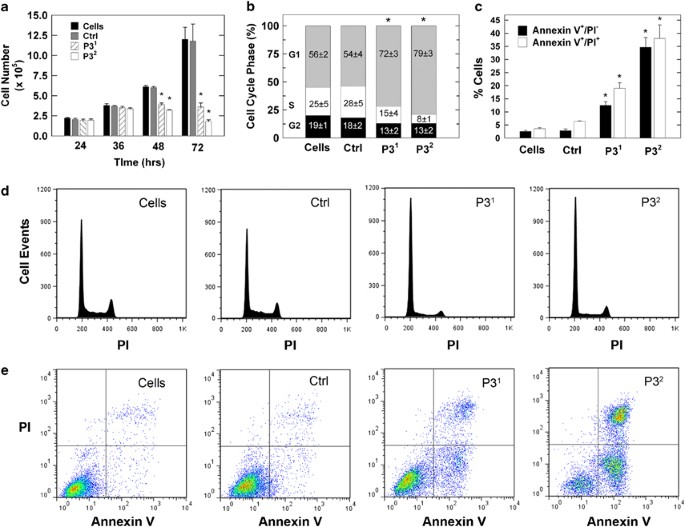
Selective inhibition of PFKFB3 suppresses cell proliferation, cell cycle progression and increases apoptosis by HeLa cells. HeLa cells were transfected with a control siRNA (Ctrl) or two PFKFB3 siRNA molecules (P31 and P32) and effects on viable cell number was assessed at 24–72 h (a), the distribution of the cells in the G1, S and G2 phases of the cell cycle after 48 h by propidium iodide (PI) staining and flow cytometry (b and d) and the presence of PI- and/or Annexin V-positive cells after 72 h by flow cytometry (c and e) were examined. *_P-_value<0.05
PFKFB3 inhibition reduces Cdk1 activity
In prior studies, we demonstrated that PFKFB3 traffics to the nucleus and produces F2,6BP that can stimulate Cdk activity.11 Based on these studies, we speculated that PFKFB3 inhibition would have the opposite effect and thus suppress Cdk activity. Initially, we transfected HeLa cells with PFKFB3 siRNA molecules and analyzed nuclear F2,6BP production and found that both PFKFB3 siRNA molecules markedly reduced the production of F2,6BP in the nuclei (Figure 3a). We then immunoprecipitated Cdk1 after PFKFB3 siRNA transfection and measured its activity using p27 as substrate. We observed a marked reduction in immunoprecipitated Cdk1 activity that correlated with the reduction in cell cycle progression and apoptosis (Figures 3b and c). We next measured the in situ Cdk1 activity by assessing the phosphorylation of serines within the Cdk1-specific (K/R)(S*)PX(K/R) motif. We observed a reduction in the phosphorylation of Cdk1 serine substrates in both the cytoplasm and nucleus after PFKFB3 siRNA transfection (Figure 3d). One such Cdk1 substrate, Cdh1, binds to and activates the APC/C ubiquitin ligase – this activation is attenuated by Cdk-mediated phosphorylation of Cdh1.14 We postulated that PFKFB3 inhibition might reduce Cdh1 phosphorylation, which in turn might increase APC/C-Cdh1 activity and decrease the stability of its substrates, which include PFKFB3 and cyclin B1. We found that transfection of HeLa cells with PFKFB3 siRNA did in fact reduce phospho-Cdh1, which in turn led to reduced levels of the APC/C-Cdh1 substrate, cyclin B1 (Figure 3e). Importantly, although each of these experiments using three different approaches clearly demonstrate that PFKFB3 expression is required to maintain Cdk1 activity, the precise mechanisms whereby the product of PFKFB3, F2,6BP, activates Cdk1 remain unknown.
Figure 3
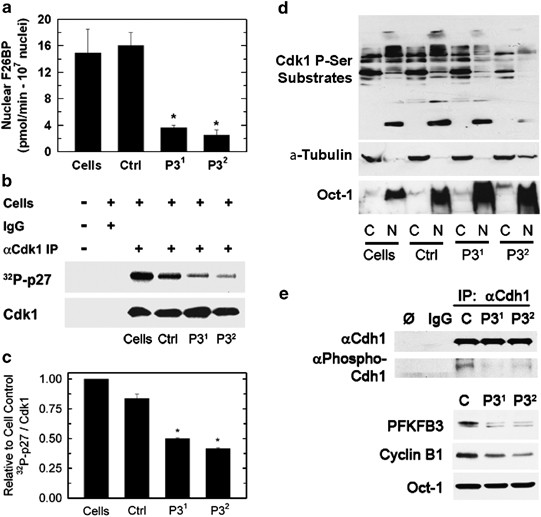
Transfection of HeLa cells with PFKFB3 siRNAs reduces nuclear F2,6BP and Cdk1 activity. HeLa cells were transfected with a control siRNA (Ctrl) or two PFKFB3 siRNA molecules (P31 and P32) and, after 48 h, nuclear F2,6BP production was quantified (a) and Cdk1 was immunoprecipitated, incubated with 32P-ATP and recombinant p27, and subjected to SDS-PAGE and western blot analysis (b) and densitometry (c). Protein lysates fractionated from the cytoplasm (C) or the nucleus (N) also were assessed for the presence of Cdk1 phosphoserine substrates by western blot analysis (d). In independent experiments, Cdh1 was immunoprecipitated and analyzed by western blot analysis using antibodies specific for total Cdh1, the Cdk1 substrates PXS*P or S*PXR/K and Oct-1 (e)
PFKFB3 inhibition results in increased nuclear and cytoplasmic p27 protein but has no effect on p57 or p21
p27 is a member of the Cip/Kip family of proteins, which are established to have regulatory roles in the cell cycle and apoptosis in part by directly suppressing Cdk1 activity.15, 16 We recently found that ectopic expression of a kinase-active PFKFB3 in the nucleus increased the Cdk-mediated phosphorylation of p27 at T187, which, in turn, resulted in a marked decrease in p27 protein as a result of ubiquitination and proteasomal degradation.11 We speculated that PFKFB3 inhibition would suppress phosphorylation of p27 at T187 and result in an increase in p27 protein concentration. Initially, we analyzed phospho-p27 protein expression in HeLa cells after PFKFB3 inhibition but did not detect any phospho-p27 in either the untreated or treated cells presumably as a result of rapid degradation of the phosphorylated p27 (data not shown). However, we did observe a marked increase in p27 protein concentration after PFKFB3 inhibition, whereas we did not observe changes in the other Cip/Kip family members, p57 and p21 (Figures 4a and b). We next confirmed that PFKFB3 inhibition also caused an increase in p27 in two unrelated transformed cell lines, HCT-116 (Figures 4c and d) and Panc-1 cells (Figures 4e and f). We performed real-time RT-PCR and found no change in p27 mRNA levels (data not shown), suggesting that the effect of PFKFB3 on p27 protein is posttranscriptional. We speculated that the stabilization of p27 would occur in the nucleus where this protein functions as a potent inhibitor of Cdk1, which is required both to promote cell cycle progression and block pro-apoptotic signals.15, 16 Surprisingly, we found that PFKFB3 inhibition caused an increase in p27 protein in the nucleus as well as the cytoplasm using fractionation and western blot analyses (Figures 4g and h) and using immunofluorescence analyses (Figure 4i).
Figure 4
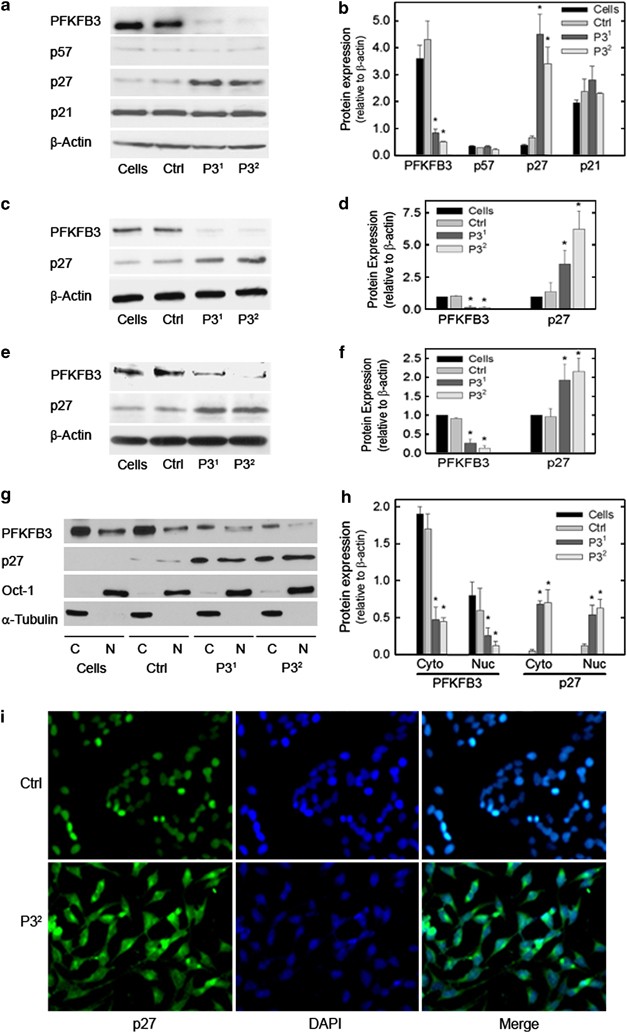
p27 protein expression is increased by selective inhibition of PFKFB3. HeLa cells were transfected with a control siRNA (Ctrl) or two PFKFB3 siRNA molecules (P31 and P32) and, after 48 h, Cip/Kip family members p57, p27 and p21 were assessed by western blot analysis (a) and densitometry (b). HCT-116 (c, d) and Panc-1 cells (e, f) were similarly transfected with the two PFKFB3 siRNA molecules (P31 and P32) and examined for p27 protein levels by western blot analysis and densitometry. HeLa cells were also separated into cytoplasmic (C) and nuclear (N) fractions, which were then assessed for PFKFB3, p27, Oct-1 (nuclear control) and a-Tubulin (cytoplasmic control) by western blot analysis (g) and densitometry (h). The Hela cells then were examined for cytoplasmic versus nuclear p27 expression by immunofluorescence using DAPI to localize the nuclei (i)
Blockade of cell cycle progression and stimulation of apoptosis by PFKFB3 inhibition is mediated by p27
Although the cytostatic and apoptotic effects of PFKFB3 inhibition have been solely attributed to the suppression of glycolysis by our group and other laboratories, we have routinely observed that the effects of PFKFB3 inhibition on viable cell number (Figure 2a) appear to be far greater than the effects on glycolysis (Figures 1e and f). Given that PFKFB3 inhibition reduces Cdk1 activity and stabilizes p27, we postulated that p27 may itself be mediating the cytostatic and pro-apoptotic effects. In order to assess the role of p27 in mediating the apoptotic and cytostatic effects of PFKFB3 inhibition, we transfected HeLa cells with control siRNA, PFKFB3 siRNA, p27 siRNA or both the PFKFB3 and p27 siRNA molecules and analyzed the effects on target protein expression, cell cycle and apoptosis after only 48 hours in order to minimize cell death and any resultant compensatory mechanisms. We found that p27 siRNA transfection completely blocked the stabilization of p27 caused by PFKFB3 inhibition, indicating that intact p27 mRNA is required to maintain continuous translation (Figures 5a and b). Importantly, whereas p27 siRNA transfection had no effect on the cell cycle or apoptosis, we found that the p27 siRNA resulted in near complete reversal of the G1/S transition block (Figures 5c and e) and induction of apoptosis (Figures 5d and f) caused by PFKFB3 siRNA transfection. In other words, suppression of p27 rescued the HeLa cells from PFKFB3 inhibition. These data demonstrate for the first time that p27 expression is required for the antiproliferative and pro-apoptotic effects of PFKFB3 inhibition.
Figure 5
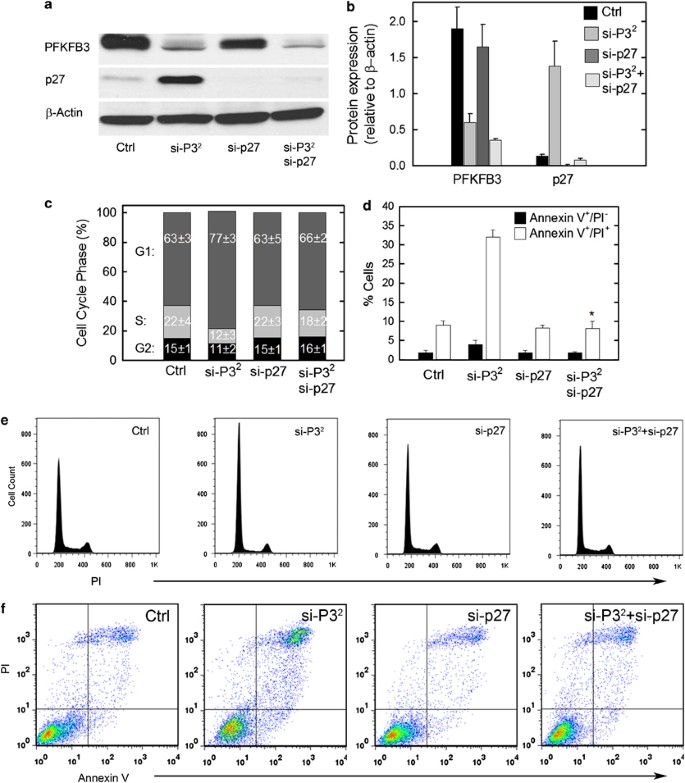
p27 is required for cell cycle block and apoptosis induced by PFKFB3 inhibition. HeLa cells were transfected with control (Ctrl), PFKFB3 siRNA (P32), p27 siRNA or both and assessed for PFKFB3, p27 or β-Actin expression by western blot analysis (a) and densitometry (b), for cell cycle phase by propidium iodide staining and flow cytometry (c and e) and PI- and/or Annexin V-staining by flow cytometry (d and f) after 48 h
The PFKFB3 inhibitor, 3PO, increases p27 protein in Lewis lung carcinoma (LLC) cells in vitro and in vivo
A small molecule antagonist of PFKFB3, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), was recently identified using a combination of computational modeling and receptor-based in silico screening.17 3PO and more potent 3PO derivatives have antitumor properties and can stimulate the apoptosis of transformed cells.12 We examined LLC cells after 3PO exposure and LLCs grown as subcutaneous tumors in syngeneic mice without and with 3PO exposure and assessed p27 expression by western blot analysis and immunohistochemistry, respectively. We observed a marked increase in p27 protein after 3PO exposure in vitro (Figures 6a and b) and in vivo (Figures 6c and d). These data indicate that p27 protein expression by tumor cells may be useful as a pharmacodynamic endpoint for incorporation into upcoming clinical trials of PFKFB3 inhibitors.
Figure 6
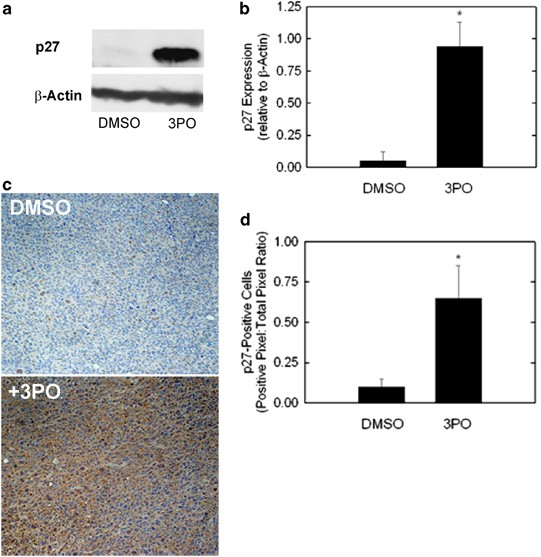
Small molecule inhibition of PFKFB3 increases p27 protein in vitro and in vivo. Lewis lung carcinoma cells were exposed to 3PO and analyzed for p27 expression by western blot (a) and densitometry (b). LLC xenograft-bearing mice were injected intraperitoneally with 3PO (0.07 mg/gm daily) and then euthanized. Three tumors were analyzed per group using immunohistochemistry for p27 (c and d)
Discussion
The data presented herein build on multiple prior studies that have suggested a potential role for PFKFB3 in cell cycle regulation and survival. For example, overexpression of fructose-2,6-bisphosphatase in transformed mink lung epithelial cells both reduced F2,6BP and decreased the number of cells that entered the S and G2/M phases of the cell cycle.18 Additionally, transfection of HeLa cells with a distinct PFKFB3-specific siRNA reduced F2,6BP and resulted in an accumulation of cells in G0/G113 and PFKFB3 siRNA has been found to suppress the proliferative effect of Pten genomic deletion in murine embryonic fibroblasts.8 Conversely, ectopic expression of the yeast PFKFB kinase domain in Schizosaccharomyces pombe increased F2,6BP and caused multiseptated and multinucleated cells with >2n DNA content.19 As PFKFB3 has been traditionally viewed as an essential regulator of glycolysis in the cytoplasm via the allosteric effects of its product, F2,6BP, on Pfk-1, the investigators of each of these four prior independent studies reasonably concluded that modulation of F2,6BP by the PFKFB family of enzymes resulted in changes in glucose metabolism, which in turn led to perturbations in the cell cycle through unidentified mechanisms.8, 13, 18, 19
We recently demonstrated that ectopically expressed PFKFB3 surprisingly localized to the nucleus and stimulated a marked increase in cell proliferation without having any effect on glucose metabolism.11 The proliferative effects of PFKFB3 were completely abrogated by mutating either the F6P-binding site or the nuclear localization residues of PFKFB3, demonstrating the requirement for nuclear delivery of F2,6BP.11 Importantly, the addition of F2,6BP to HeLa cell lysates increased the phosphorylation of the Cdk-specific Thr-187 site of p27, which marks p27 for proteasomal degradation and ectopic expression of PFKFB3 decreased p27 protein levels.11 p27 is a nonclassical tumor suppressor and potent inhibitor of Cdk activity and the cell cycle.15, 16, 20, 21, 22, 23, 24 Specifically, p27 can physically bind to and block the activity of the Cdks, leading to cell cycle inhibition and has the ability to induce apoptosis in several transformed cells when ectopically expressed.15 Whereas recent studies have identified Cdk1 as an important in vivo target of p27, p27 is also a substrate for Cdk1 and Cdk2 and phosphorylation of p27 at T187 by Cdk1 and Cdk2 targets p27 to the proteasome for degradation.24, 25 Accordingly, the regulation of Cdk1 and p27 is intimately linked and these two proteins can dictate cell proliferation versus cell death.
In the current study, we have examined the specific role of endogenous PFKFB3 protein expression and F2,6BP in regulating Cdk1 activity, p27 expression, the cell cycle and resistance to apoptosis. We demonstrate that PFKFB3 inhibition reduces the phosphorylation of p27 by Cdk1 causing a marked increase in p27 protein, which then results in both a G1/S block and an increase in apoptosis. We confirm the essential role of p27 by demonstrating that silencing of p27 expression attenuates the cell cycle block and apoptosis caused by PFKFB3 inhibition. These new findings coupled to the observations made in our prior study11 support a model whereby nuclear PFKFB3-synthesized F2,6BP activates Cdk1 to phosphorylate the Thr-187 site of p27, resulting in decreased p27, acceleration of cell cycle progression at the G1/S phase transition and protection against apoptosis (Figure 7). Based on these studies, we thus conclude that PFKFB3 functions not only as a regulator of glucose metabolism but also as a direct regulator of the cell cycle.
Figure 7
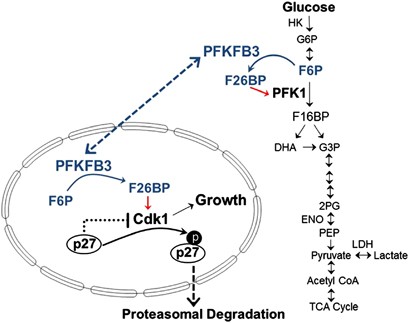
PFKFB3 synthesizes F2,6BP, which in turn activates PFK-1 and glycolysis in the cytosol and Cdk1 in the nucleus. Taken together, the data presented in the initial report of the functions of nuclear PFKFB3 and this article support a model whereby PFKFB3 in the cytosol regulates glucose metabolism, whereas PFKFB3 in the nucleus regulates Cdk1 activity and p27 protein levels. Both glucose metabolism and Cdk1 activity are required for growth and resistance to apoptosis
Importantly, the observation that selective inhibition of p27 was able to completely rescue HeLa cells from PFKFB3 inhibition suggests that suppression of Cdk1 is the key event that leads to the antiproliferative and pro-apoptotic effects of PFKFB3 inhibition in HeLa cells. Although a functional glucose deprivation can have very similar effects, we believe that targeting PFKFB3 is analogous to simultaneously hitting the brakes (i.e. p27) and siphoning a portion of the gasoline (i.e. glucose metabolism) in an automobile. The vehicle will stop even if some gasoline is available (note that PFKFB3 inhibition reduced glycolysis only by ∼50%, Figure 1f) but will go if the brake is released (e.g. via p27 siRNA). However, loss of p27 expression through epigenetic changes could leave the brake off and oxygen- and glucose-poor microenvironments could result in sensitivity to effects of PFKFB3 inhibition on glucose metabolism. Given these two scenarios, we suspect that the antitumor effects of PFKFB3 inhibition are likely due to the suppression of a combination of both the Cdk1 activity and glucose metabolism. Additionally, we predict that the relative significance of regulation of the cell cycle versus glucose metabolism likely depends on the intracellular localization of PFKFB3 in the nucleus and cytosol.
The observation that the PFKFB3 inhibitor, 3PO, similarly results in increased p27 protein in LLC cells in vitro and tumors in vivo indicates that PFKFB3 inhibitors may have antitumor effects that depend on both their established ability to reduce glucose metabolism and their ability to disrupt the cell cycle and stimulate apoptosis. At a minimum, these data indicate that p27 protein levels may be useful as a biomarker in biopsied tumor tissues to correlate objective responses with on-target effects. More importantly, these findings open avenues for the rational development of combination approaches that simultaneously disrupt the functions of the Cdks and PFKFB3.
Several previous studies have demonstrated that the E3 ubiquitin ligase APC/C-Cdh1 simultaneously regulates not only the cell cycle but also metabolism by selectively targeting key regulatory enzymes, including PFKFB3, for proteasomal degradation.26, 27, 28 Our data indicate that, in fact, APC/C-Cdh1 may be regulating the stability of PFKFB3 not just to modulate glucose metabolism but also to regulate Cdk1 and the cell cycle. Given that: (i) PFKFB3 is an APC/C-Cdh1 substrate;28 (ii) Cdh1 phosphorylation by Cdks has been found to inhibit its ability to activate the ubiquitin ligase activity of APC/C;14 and (iii) Cdk1-promoted Cdh1 inhibition causes PFKFB3 stabilization,29 we speculate that the stimulatory effects of F2,6BP on Cdk1 activity may result in phosphorylation of Cdh1, which in turn may suppress APC/C-Cdh1 activity and increase PFKFB3. In support of this potential positive feedback loop, we found that selective PFKFB3 inhibition suppressed the Cdk-mediated phosphorylation of Cdh1, which in turn resulted in reduced cyclin B1 protein, an established target of APC/C-Cdh1 that is not directly inhibited by the PFKFB3 siRNA. Based on these results, we also postulated that the observed stabilization of p27 by PFKFB3 inhibition may be partly due to decreased APC/C-Cdh1-mediated degradation of Skp2 which would result in reduced SCF ubiquitin ligase-mediated degradation of p27.30 However, we did not observe any reduction in Skp2 protein levels after PFKFB3 siRNA transfection (data not shown). Future studies will be directed at interrogating the specific roles of both APC/C-Cdh1 and SCF ubiquitin ligases in the modulation of the newly established functions of PFKFB3 and F26BP in directly regulating cell cycle progression via Cdks and p27.
Materials and Methods
Cell culture
HeLa, HCT-116, PANC-1 and LLC cells were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Clontech, Mountain View, CA, USA) under 5% CO2 at 37 °C.
SiRNA transfections
Transfection of siRNA molecules was carried out using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions. Cells were 40–50% confluent at the time of transfection. The following siRNAs for PFKFB3 and p27 were used: PFKFB3 siRNA#1 (termed as P31), 5′-GCUGUGAAGCAGUACAGCUCCUAC-3′ (Invitrogen, #PFKFB3HSS107862); PFKFB3 siRNA#2 (termed as P32), 5′-GCCGCAUCGUGUACUACCUGAUGAA-3′ (Invitrogen, #PFKFB3HSS107860); p27 siRNA, 5′-GCCAGCGCAAGUGGAAUUUCGAUUU-3′ (Invitrogen, #CDKN1BHSS101741). The universal control siRNA (Invitrogen #12935-112) that has no homology to any sequence in the human genome and the identical GC content of the two PFKFB3 siRNA molecules was used as a control.
Real-time RT-PCR
RNA isolation and cDNA synthesis were performed using commercial kits (Qiagen, Germantown, MD, USA) following manufacturer’s instructions. PFKFB3 and p27 mRNA expression was determined by Syber Green-based real-time PCR analysis of cDNAs synthesized. All primers were obtained from Integrated DNA Technologies (Coralville, IA, USA). The primers used were as follows: PFKFB3, 5′-CAGTTGTGGCCTCCAATATC-3′ (forward) and 5′-GGCTTCATAGCAACTGATCC-3′ (reverse); p27, 5′-ACTTGGAGAAGCACTGCAGAGACA-3′ (forward) and 5′-TCTGTAGTAGAACTCGGGCAAGCTG-3′ (reverse); _β_-actin, 5′-CCAACCGCGAGAAGATGACC-3′ (forward) and 5′-GGAGTCCATCACGATGCCAG- 3′ (reverse).
Fluorescence microscopy
Transfected cells grown on coverslips were fixed in paraformaldehyde and processed as described previously.31 Incubation with Alexa Fluor 488-labeled anti-p27 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was performed overnight. Immunofluorescence images were acquired with Zeiss Axiovert 200 microscope.
Cytoplasmic and nuclear fractionation
Nuclear and cytosolic fractions were extracted using NE-PER Nuclear and Cytoplasmic Extractions Kit (Pierce Biotechnology, Rockford, IL, USA). Extraction procedure was done according to the manufacturer’s protocol.
Small molecule inhibition of PFKFB3
LLC cells were plated at a density of 1 × 105 cells/well in 6-well plates. Twenty-four hours later, either DMSO as vehicle control or 3PO (Chembridge, San Diego, CA, USA) was added at a final concentration of 30 _μ_M. Forty-eight hours later, cells were lysed and subjected to western blot analysis as described below for p27 expression.
Western blot analyses
For preparation of total cell lysates, cells were washed in cold PBS and lysed with a lysis buffer (50 mM Tris-HCL, pH 7.5, 1 mM EDTA, 150 mM NaCI, 5 mM MgCI2, 0.5% NP-40 and 0.5% Triton-X-100) containing protease and phosphatase inhibitors (Sigma, St. Louis, MO, USA). Samples were separated through a 12.5% SDS-PAGE gel under reducing conditions, and proteins were then transferred to Immobilon P membranes. Rabbit polyclonal PFKFB3 antibody was obtained from Proteintech (Chicago, IL, USA; #13763-1-AP). Mouse monoclonal anti-_α_-tubulin was purchased from NeoMarkers (Fremont, CA, USA). Anti-Oct-1 was obtained from Santa Cruz Biotechnology. Rabbit polyclonals anti-Cdk1, p21, p27, p57, phospho(Ser) CDKs substrate, and HRP-conjugated goat anti-rabbit and anti-mouse IgG antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody specific for phospho-p27 (T187) was obtained from Invitrogen. Mouse monoclonal anti-_β_-actin was purchased from Sigma. For detection of immunoreactive bands, ECL and ECL Prime were used (Amersham, Pisctaway, NY, USA).
F2,6BP assay
Intracellular F2,6BP concentration was determined as previously described.32 Briefly, cells were centrifuged at 200 × g, resuspended in 20 volumes of 0.05 N NaOH and then 1 volume of 0.1 N NaOH to obtain a pH >11, vortexed for 10 s, incubated at 80 °C for 5 min and cooled in an ice bath. Cell extracts were neutralized to pH 7.2 with ice-cold acetic acid in the presence of 20 mM Hepes. Samples were incubated at 25 °C for 2 min in the following assay mixture: 50 mM Tris, 2 mM Mg+2, 1 mM F6P, 0.15 mM NAD, 10U/l PPi-dependent PFK-1, 0.45 kU/l aldolase, 5 kU/l triosephosphate isomerase and 1.7 kU/l glycerol-3-phosphate dehydrogenase (Sigma). In total, 0.5 mM pyrophosphate was added and the rate of change in absorbance (OD=339 nm) per min was followed for 5 min. F2,6BP was calculated based on a calibration curve produced by measuring 0.1 to 1 pmol of F2,6BP (Sigma) and normalized to total cellular protein.
2-[1-14C]-Deoxy-d-glucose uptake assay
HeLa cells were transfected with PFKFB3 siRNA as described above. Forty-eight hours later, cells were washed twice with PBS and incubated in glucose-free media for 30 min. Cells were then treated with 25 _μ_l of 14C-deoxy-glucose (0.1 _μ_Ci/_μ_l) for 1 h, washed once with ice-cold glucose-free DMEM and twice with ice-cold PBS. Cells were lysed in 500 _μ_l 0.5% SDS. Four hundred microliters of lysate was added to 5 ml Microscint 40 scintillation fluid (Perkin-Elmer, Akron, OH, USA) and counts were measured on a Tri-Carb 2910 liquid scintillation analyzer (Perkin-Elmer). Remaining lysate was quantitated with the BCA Assay (Pierce) per manufacturer’s instructions. Counts were normalized to protein concentration.
Glycolysis assay
HeLa cells were transfected with PFKFB3 siRNA as described above. Forty-eight hours later, cells were washed twice with PBS and incubated in Krebs buffer containing 1 mM nonradioactive glucose and 1 _μ_Ci [5-3H]glucose and incubated for 60 min in 5% CO2 at 37 °C. Hundred microliter aliquots were added to 50 _μ_l 0.2 N HCl in open tubes that were placed upright in scintillation vials containing 1 ml H2O. The vials were sealed and the [3H]H2O produced by glycolysis through enolase was allowed to equilibrate with the H2O in the open tubes for 24 h at room temperature. The amounts of 3H in both compartments were determined separately and compared with [3H]H2O and [5-3H]glucose standards.
Flow cytometry
Cells were washed with PBS and stained with Annexin V and Propidium Iodide (BD Pharmingen, San Diego, CA, USA). Fluorescence was measured using a FACSCalibur (BD Biosciences, San Jose, CA, USA) and analyzed using FloJo (Tree Star, Ashland, OR, USA). Annexin V+/PI+ (late apoptotic) and Annexin V+/PI− (early apoptotic) cells were quantified by the frequency of fluorescently labeled cells and statistical significance was assessed by the two-sample _t_-test (independent variable).
In vitro 6-phosphofructo-2-kinase assay on purified nuclei
Nuclei for in vitro 6-phosphofructo-2-kinase activity were isolated from HeLa cells using a Nuclei Isolation Kit (Sigma, Cat#NUC-101) following the manufacturer’s instructions. The purity of nuclei was confirmed to be over 95% based on trypan blue staining. The following assay was developed to measure the generation of F2,6BP by relatively intact pure nuclei. Approximately 1 × 107 nuclei was incubated at 30 °C for 10, 20, and 40 min while shaking in a 200 ul kinase buffer containing 100 mM Tris-HCl (pH 7.5), 0.5 mM DTT, 0.1 mM EDTA, 5 mM ATP, 1 mM Fru-6-P, 5 mM potassium phosphate (KH2PO4) and 10 mM MgCl2. Aliquots from supernatant were used to measure F2,6BP production (see F2,6BP assay above). No substrate (F6P) control was included to correct for background signal.
Cdk1 kinase assay
HeLa cells (1 × 106) were transfected with two separate PFKFB3 siRNA species for 48 h. Post transfection, cell lysates were collected and standardized for protein content across all samples. Cdk1 was immunoprecipitated from lysates using anti-Cdk1 (C19, Santa Cruz Biotechnology) with rotation at room temperature for 1 h. Pre washed protein-G-sepharose (Dynabeads, Invitrogen) then was added to the IP samples for an additional 30 min at room temperature and subsequently washed 3 × with binding buffer. Immunoprecipitates were resuspended in a kinase reaction mixture containing 5 _μ_g recombinant p27, 0.5 mM cold ATP, and 0.2 _μ_Ci 33P-ATP and incubated at 30 °C for 10 min. Reactions were removed from the sepharose beads and stopped by addition of 2 × Laemmli buffer. Proteins were separated via SDS-PAGE, and 33P-p27 was visualized by exposure to a phosphorscreen. Subsequently, immunoprecipitated CDK1 was determined by direct addition of Laemmli buffer to the beads and subjected to SDS-PAGE and western blot analysis using anti-Cdk1 (Cell Signaling Technology).
Cdh1 Immunoprecipitation and phosphorylation
Nuclear lysates were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) following the manufacturer’s instructions. One hundred and fifty microgram of nuclear lysates were incubated overnight at 4 °C with a Cdh1 antibody (Abcam, Cambridge, MA, USA; ab3242) or IgG isotope control in RIPA buffer supplemented with protease and phosphatase inhibitors. Dynabeads Protein G (Life Technologies, Carlsbad, CA, USA) was added and incubated for 1 h with rotation at 4 °C. Proteins were eluted in SDS sample buffer by incubating at 60 °C for 30 min. Proteins in the resulting supernatant were separated on a 10% Mini PROTEAN TGX gels (Bio-Rad, Hercules, CA, USA). The membranes were blocked and immunoblotted with a phospho-CDK substrate antibody (Cell Signaling Technology; 34B2) against phosphoserine in a PXS*P or S*PXR/K motif.
Immunohistochemistry
Five-micrometer sections of formalin-fixed and paraffin-embedded tumor tissue were mounted on charged glass slides and dried at 58 °C for 60 min. Slides were first deparaffinized with xylene then epitope retrieval carried out using citrate buffer in a 2100 Retriever pressure cooker (PickCell Laboratories, San Jose, CA, USA). The sections were blocked with 10% goat serum for 1 h, then incubated with primary antibody (anti-p27, 1 : 50, Abcam (ab7961)) overnight, followed by an HRP-linked goat anti-rabbit secondary antibody (1 : 300, Pierce Biotechnology). The sections were developed with 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories, Burlingame, CA, USA) for 2 min and nuclei counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO, USA) for 2 min. PBS washes were performed between all steps. The slides were dehydrated in graded alcohols (100%, 95% and 80% ethanol (vol/vol) in H2O), cleared in xylene and coverslips attached with Permount (Fisher Scientific, Waltham, MA, USA). Slides were scanned using a ScanScope XT Digital Slide Scanner (Aperio, Vista, CA, USA) and data were analyzed with the positive pixel count algorithm (ImageScope, Aperio).
Statistics
In order to determine whether the difference in the mean values of the two examined groups is greater than would be expected by chance, we employed SigmaPlot 12.3 to conduct two tailed _t_-tests and reported the resultant _P_-values.
Conclusions
We conclude that PFKFB3 functions to activate glucose metabolism, promote cell cycle progression and prevent apoptosis in transformed cells. We expect that the characterization of the precise regulatory network that allows a single small molecule, F2,6BP, to couple glucose metabolism with proliferation and survival will result in the identification of multiple novel molecular targets that will prove useful for the development of antineoplastic agents.
Abbreviations
PFKFB1-4:
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases
Cdk:
cyclin-dependent kinase
T187:
threonine 187
F2,6BP:
fructose-2,6-bisphosphate
Pfk-1:
6-phosphofructo-1-kinase
F6P:
fructose 6-phosphate
3PO:
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
LLC:
Lewis lung carcinoma
DMSO:
dimethyl sulfoxide
References
- Yalcin A, Telang S, Clem B, Chesney J . Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol 2009; 86: 174–179.
Article CAS Google Scholar - Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y et al. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci USA 1999; 96: 3047–3052.
Article CAS Google Scholar - Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N et al. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem (Tokyo). 1997; 122: 122–128.
Article CAS Google Scholar - Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res 2002; 62: 5881–5887.
CAS PubMed Google Scholar - Kessler R, Eschrich K . Splice isoforms of ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in human brain. Brain Res Mol Brain Res 2001; 87: 190–195.
Article CAS Google Scholar - Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem 2004; 279: 53562–53570.
Article CAS Google Scholar - Manes NP, El-Maghrabi MR . The kinase activity of human brain 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase is regulated via inhibition by phosphoenolpyruvate. Arch Biochem Biophys 2005; 438: 125–136.
Article CAS Google Scholar - Cordero-Espinoza L, Hagen T . Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (pten)-deficient cells. J Biol Chem 2013; 288: 36020–36028.
Article CAS Google Scholar - Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149: 49–62.
- Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, Eaton JW et al. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 2006; 25: 7225–7234.
Article CAS Google Scholar - Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 2009; 284: 24223–24232.
Article CAS Google Scholar - Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr 2nd DA et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 2013; 12: 1461–1470.
Article CAS Google Scholar - Calvo MN, Bartrons R, Castano E, Perales JC, Navarro-Sabate A, Manzano A . PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett 2006; 580: 3308–3314.
Article CAS Google Scholar - Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolanos JP, Almeida A . Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J 2008; 27: 2736–2745.
Article CAS Google Scholar - Wang X, Gorospe M, Huang Y, Holbrook NJ . p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene 1997; 15: 2991–2997.
Article CAS Google Scholar - Besson A, Dowdy SF, Roberts JM . CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008; 14: 159–169.
Article CAS Google Scholar - Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 2008; 7: 110–120.
Article CAS Google Scholar - Perez JX, Roig T, Manzano A, Dalmau M, Boada J, Ventura F et al. Overexpression of fructose 2,6-bisphosphatase decreases glycolysis and delays cell cycle progression. Am J Physiol Cell Physiol 2000; 279: C1359–C1365.
Article CAS Google Scholar - Fernandez de Mattos S, Alemany V, Aligue R, Tauler A . Increase in Fru-2,6-P(2) levels results in altered cell division in Schizosaccharomyces pombe. Biochim Biophys Acta 2008; 1783: 144–152.
Article CAS Google Scholar - Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F . p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle 2007; 6: 1053–1061.
Article CAS Google Scholar - Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994; 78: 59–66.
Article CAS Google Scholar - Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ . The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998; 396: 177–180.
Article CAS Google Scholar - Blain SW, Scher HI, Cordon-Cardo C, Koff A . p27 as a target for cancer therapeutics. Cancer Cell 2003; 3: 111–115.
Article CAS Google Scholar - Kaldis P . Another piece of the p27Kip1 puzzle. Cell 2007; 128: 241–244.
Article CAS Google Scholar - Aleem E, Kiyokawa H, Kaldis P . Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol 2005; 7: 831–836.
Article CAS Google Scholar - Almeida A, Bolanos JP, Moncada S . E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci USA 2010; 107: 738–741.
Article CAS Google Scholar - Bolanos JP . Adapting glycolysis to cancer cell proliferation: the MAPK pathway focuses on PFKFB3. Biochem J 2013; 452: e7–e9.
Article CAS Google Scholar - Herrero-Mendez A, Almeida A, Fernandez E, Maestre C, Moncada S, Bolanos JP . The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 2009; 11: 747–752.
Article CAS Google Scholar - Rodriguez-Rodriguez P, Fernandez E, Almeida A, Bolanos JP . Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ 2012; 19: 1582–1589.
Article CAS Google Scholar - Cuende J, Moreno S, Bolanos JP, Almeida A . Retinoic acid downregulates Rae1 leading to APC(Cdh1) activation and neuroblastoma SH-SY5Y differentiation. Oncogene 2008; 27: 3339–3344.
Article CAS Google Scholar - Fry AM, Meraldi P, Nigg EA . A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. Embo J 1998; 17: 470–481.
Article CAS Google Scholar - Van Schaftingen E, Lederer B, Bartrons R, Hers HG . A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur J Biochem 1982; 129: 191–195.
Article CAS Google Scholar
Acknowledgements
We thank the following funding agencies for their support of these studies: AY: European Union Research Executive Agency, #PIRG06-GA-2009-256523, YIF and JO: CDMRP Post-Doctoral Fellowships; ST: NCI 1R01CA140991 and ACS RSG-10-021-01CNE; and JC: NCI 1R01CA149438.
Author information
Authors and Affiliations
- Departments of Medicine (Hematology/Oncology) and Biochemistry and Molecular Biology, University of Louisville, James Graham Brown Cancer Center, Louisville, 40202, KY, USA
A Yalcin, B F Clem, Y Imbert-Fernandez, J O'Neal, A C Klarer, A L Clem, S Telang & J Chesney - Department of Biochemistry, School of Veterinary Medicine, Uludag University, Bursa, Turkey
A Yalcin & S C Ozcan - Department of Histology and Embryology, School of Veterinary Medicine, Uludag University, Bursa, Turkey
S Peker
Authors
- A Yalcin
- B F Clem
- Y Imbert-Fernandez
- S C Ozcan
- S Peker
- J O'Neal
- A C Klarer
- A L Clem
- S Telang
- J Chesney
Corresponding author
Correspondence toJ Chesney.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yalcin, A., Clem, B., Imbert-Fernandez, Y. et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27.Cell Death Dis 5, e1337 (2014). https://doi.org/10.1038/cddis.2014.292
- Received: 27 January 2014
- Revised: 19 April 2014
- Accepted: 29 April 2014
- Published: 17 July 2014
- Issue Date: July 2014
- DOI: https://doi.org/10.1038/cddis.2014.292