Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia (original) (raw)
Introduction
SATB2 is a gene located at the chromosomal region 2q33.1 encoding a highly conserved DNA-binding protein known to have a role in craniofacial and neuronal development.1, 2, 3, 4, 5, 6 Recently, Döcker et al7 provided a summary of patients harboring an alteration of this gene in this Journal. They delineated the SATB2 phenotype by comparing four patients with deletions restricted to SATB2,8, 9 two patients with the same heterozygous nonsense variant (c.715C>T, p.R239*)7, 10 and preliminary information on the patient of this report with an intragenic SATB2 duplication, presented at that time in abstract form.11 They proposed a clinically recognizable SAS (_SATB2_-associated syndrome), characterized by severe intellectual disability (ID) with no or only limited speech, behavioral problems and abnormalities in craniofacial patterning, namely micrognathia, cleft or high-arched palate, and abnormalities of the teeth such as oligodontia and/or misshaped and crowded teeth.7 In the meantime, Rainger et al12 have published about three unrelated individuals with translocation breakpoints 3′ of SATB2 and a comparable phenotype to that in patients with SATB2 mutations, suggesting the action of _cis_-regulatory elements in this region as a further pathomechanism. The patient of this report represents the first case of an intragenic SATB2 duplication and has not been described with sufficient clinical and molecular data so far. Therefore, in reaction to the article by Döcker et al,7 we would like to add a detailed description of the patient’s phenotype and genotype, correcting hereby the information given in the paper by Döcker et al,7 and adding to the knowledge on molecular mechanisms leading to SAS.
Case report
This 10-year-old girl was the only child of German, non-consanguineous parents. The pregnancy was the result of an intracytoplasmatic sperm injection procedure performed because of a mild cystic fibrosis with CBAVD in the father. Apart from that the family history was unremarkable. The girl was born at term spontaneously following an uneventful pregnancy; birth weight was 3760 g (75th centile), length 55 cm (90–95th centile) and occipito-frontal circumference (OFC) 35 cm (50th centile). The neonatal period was inconspicuous. At the age of 33 months, she was first referred for genetic evaluation because of global developmental delay, no speech development, sleeping problems and feeding difficulties. Conventional karyotyping, FMR1 analysis and investigations for inborn errors of metabolism showed all normal results. MRI of the brain at the age of 2 years, showed a pathologic myelinization with increased signal intensity in the right parieto-occipital region. At the age of 10 years and 3 months, she was seen again in the clinic of Human Genetics. She showed moderate to severe ID but a formal testing had not been performed so far, and she attended a school for disabled children. She had a nearly absent speech; her vocabulary consisted of about 10 words. Her speech perception was much better and she was able to communicate by gestures. Her growth was in the low normal range: weight 28.3 kg (10–25th centile), height 136.5 cm (10th centile) and OFC 52.5 cm (50th centile). On physical examination, a narrow jaw with crowded teeth and high palate were noted (no cleft palate, as described before7). Hypodontia was suspected (Figure 1c) but could not be ensured by the radiograms taken so far. She had short palpebral fissures, a broad nose with broad nasal bridge, bulbous tip and thick columella, short hands (length 14 cm, 3rd centile), mildly broad thumbs and big toes (Figure 1a, b and d). No other skeletal anomalies were noticed. In contrast to the previous description by Döcker et al,7 the girl had no challenging but a very friendly and outgoing character. Sleeping problems and feeding difficulties had improved. Seizures, other neurological problems or visceral malformations were not present.
Figure 1
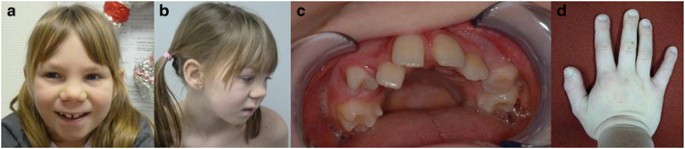
Clinical photographs of the patient. (a, b) Patient at the age of 10 years showing mild dysmorphic features, (c) upper jaw of the patient with suspected hypodontia and (d) right hand of the patient with mildly broad thumbs.
Materials and methods
DNA and RNA isolation
Genomic DNA was isolated from peripheral blood using a salting out protocol according to Miller et al.13 Total RNA was extracted from the peripheral blood of the patient, her parents and an unrelated control person using a phenol–chloroform method as described by Chomczynski and Sacchi.14
Microarray-analysis
Oligo/SNP-Array analysis was performed on the genomic DNA of the patient using the Affymetrix CytoScan HD array according to the manufacturer’s instructions. Arrays were scanned with the AffymetrixGeneChip Scanner 3000 7G and genotypes were analyzed with Affymetrix Chromosome Analysis Suite software version 1.2 and Annotation Net Affx Build 32 (Affymetrix, Santa Clara, CA, USA). Interpretation was based on human reference sequence (GRCH37/hg19, February 2009). The patient data were submitted to DECIPHER database (https://decipher.sanger.ac.uk/, identification number IHG290894).
Multiplex ligation-dependent probe amplification
Customized multiplex ligation-dependent probe amplification (MLPA) analysis was performed using MRC Holland’s reference mix P200-A1 together with synthetic probes located in exon 1, intron 2, intron 3 and exon 4 of the SATB2 gene (NG_016976.1, exon numbering is according to Ensemble transcript ENST00000417098). Probe sequences were obtained using the MLPA probe design tool (H-MAPD)15 following the recommendations described in MRC Holland’s probe design guidelines (MRC Holland, Amsterdam, The Netherlands). Oligonucleotides were purchased from Sigma-Aldrich (München, Germany). Synthetic probe mixes were prepared in TE buffer (10 mM Tris-HCL, pH 8.0; 1 mM EDTA) following MRC Holland’s recommendations with minor modifications (available on request). The MLPA analysis was carried out on the genomic DNA of the patient and her parents as described by Schouten et al.16
Fine mapping of duplication breakpoints
Standard PCRs on the genomic DNA of the patient were carried out with primers flanking the predicted duplication breakpoint regions using the Invitrogen Platinum Taq polymerase system (Invitrogen/Life Technologies, Darmstadt, Germany). PCR products were cleaned up with Exonuclease I and Fast AP alkaline phosphatase (Thermo Fisher Scientific, St Leon Rot, Germany). Sanger sequencing was performed as mentioned below.
cDNA analysis
cDNA of the patient, her parents and a healthy, unrelated control individual was obtained through reverse transcription of RNA by using Superscript II reverse transcriptase (Life Technologies) according to the manufacturer’s instructions. The cDNA was amplified using the Invitrogen Platinum Taq polymerase system (Invitrogen/Life Technologies) with a forward primer located in exon 2 and a reverse primer located in exon 4 of the SATB2 gene. PCR products were separated by electrophoresis on a 2% agarose gel and cDNA bands representing the wild-type and the duplicated allele were extracted using the peqGOLD Gel Extraction kit (PEQLAB Biotechnologie, Erlangen, Germany). Sanger sequencing of the extracted fragments was performed as mentioned below.
Mutation screening
The coding region of SATB2 (exons 2–11, transcript NM_001172509.1/ENST00000417098) including exon/intron boundaries was amplified from the genomic DNA of the patient. Sanger sequencing was performed as mentioned below.
Sequence analysis
Sanger sequencing was performed using version 1.1 of Big Dye terminator cycle sequencing kit (Applied Biosystems/Life Technologies, Darmstadt, Germany). Sequence products were analyzed on a 3130xl Genetic Analyzer (Applied Biosystems/Life Technologies). Primers and amplification procedures are available on request.
Results
Molecular karyotyping of the patient showed an 84-kb duplication within chromosomal region 2q33.1 (arr[hg19] 2q33.1(200,256,583–200,340,204) × 3) encompassing a part of the SATB2 gene (Figure 2a). In addition, a 900-kb duplication in chromosomal region 16q22.3 was detected that did not seem to be related to the patient’s phenotype. Subsequent MLPA analysis of the girl and her parents confirmed a de novo duplication of exon 3 of the SATB2 gene in the patient (exon numbering according to ENST00000417098). Sequence analysis of genomic DNA using primers flanking the predicted duplication breakpoints revealed that the duplication is in tandem with no additional sequence changes (Figure 2d). Furthermore, on the basis of this analysis the precise position of the duplication could be narrowed down to 200 256 546–200 310 885 bp (chr2:g.200256546_200310885 dup/NM_001172509.1: c.169+9707_347-10003dup) with a duplication size of 54 kb. Reverse transcription PCR analysis with cDNA primers flanking the duplicated exon 3 resulted in a 314-bp wild-type fragment in the parents and one healthy control individual, and two additional fragments of ∼ 500 bp and 600 bp in the patient (Figure 2f). Subsequent Sanger sequencing of the gel-extracted cDNA fragments showed that the 500-bp fragment represented the transcript of the allele with the tandem duplication of exon 3 in frame. This is consistent with coexpression of transcripts with a duplicated exon 3 and wild-type transcripts in the patient. The 600-bp fragment could not be extracted from the gel and thus further characterization was not possible.
Figure 2
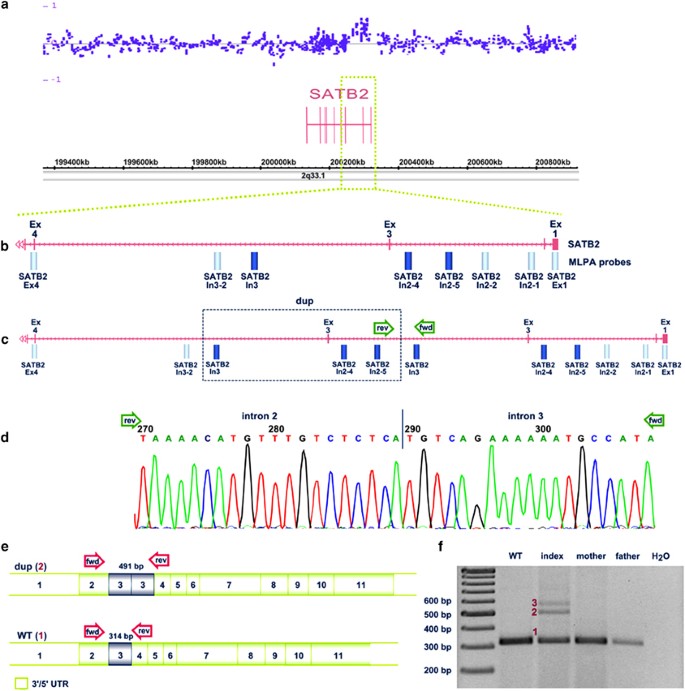
Molecular genetic investigations. (a) Array profile of the patient representing the duplicated region within SATB2; (b) zoom into the SATB2 region of interest showing localization of MLPA probes (dark bars: duplicated probes; light bars: normal probes); (c) schematic view of the duplication (box) within SATB2 and localization of primer pairs for duplication breakpoint mapping (arrows); (d) sequence analysis of the duplication breakpoint on genomic DNA level. (e) cDNA: schematic view of wild-type and aberrant SATB2 transcripts with localization of cDNA primer pairs (arrows). (f) cDNA analysis showing the 314-bp wild-type fragment (1) in the parents and a healthy control (WT) and two additional fragments of ∼500 bp (2) and 600 bp (3) in the patient. Exon numbering is according to Ensemble transcript ENST00000417098. bp, basepair; dup, duplication; fwd, forward; rev, reverse; UTR, untranslated region; WT, wild-type.
To exclude the presence of a pathogenic variant on the second allele Sanger sequence analysis of the 10 coding exons (exons 2–11) and the exon/intron boundaries of SATB2 was performed and did not show any causative variants.
Conclusion
Here we present a detailed clinical and molecular description of the first case with an intragenic SATB2 duplication leading to a similar phenotype as described for patients with intragenic SATB2 deletions or a heterozygous SATB2 nonsense mutation.6, 7, 8, 9, 10 The patient presented with ID, nearly absent speech and suspected hypodontia, the characteristic features of SAS. By molecular genetic investigations we were able to confirm a tandem duplication of exon 3 with different coexpressed transcripts of SATB2. We hypothesize that the aberrant transcripts lead to functionally impaired or nonfunctional proteins and consequently to haploinsufficiency of SATB2. Sequencing of the entire SATB2 gene revealed no additional sequence alteration, which is in line with an autosomal dominant entity of SAS, caused by haploinsufficiency of SATB2. This case provides further insight into the spectrum of the molecular mechanisms leading to SAS.
References
- Sheehan-Rooney K, Palinkasova B, Eberhart JK, Dixon MJ : A cross-species analysis of Satb2 expression suggests deep conservation across vertebrate lineages. Dev Dyn 2010; 239: 3481–3491.
Article CAS Google Scholar - Dobreva G, Chahrour M, Dautzenberg M et al: SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 2006; 125: 971–986.
Article CAS Google Scholar - Alcamo EA, Chirivella L, Dautzenberg M et al: Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 2008; 57: 364–377.
Article CAS Google Scholar - Britanova O, Depew MJ, Schwark M et al: Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet 2006; 79: 668–678.
Article CAS Google Scholar - Leyva-Diaz E, Lopez-Bendito G : In and out from the cortex: development of major forebrain connections. Neuroscience 2013; 254: 26–44.
Article CAS Google Scholar - Zhao X, Qu Z, Tickner J, Xu J, Dai K, Zhang X : The role of SATB2 in skeletogenesis and human disease. Cytokine Growth Factor Rev 2013; 25: 35–44.
Article Google Scholar - Döcker D, Schubach M, Menzel M et al: Further delineation of the SATB2 phenotype. Eur J Hum Genet 2013; 22: 1034–1039.
Article Google Scholar - Rosenfeld JA, Ballif BC, Lucas A et al: Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS One 2009; 4: e6568.
Article Google Scholar - Balasubramanian M, Smith K, Basel-Vanagaite L et al: Case series: 2q33.1 microdeletion syndrome—further delineation of the phenotype. J Med Genet 2011; 48: 290–298.
Article CAS Google Scholar - Leoyklang P, Suphapeetiporn K, Siriwan P et al: Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Human Mutat 2007; 28: 732–738.
Article CAS Google Scholar - Kaiser AS, Maas B, Wolff A et al: Girl with developmental delay, nearly absent speech and oligodontia: First case of an intragenic SATB2 duplication. Medizinische Genetik 2013; 25: 125.
Google Scholar - Rainger JK, Bhatia S, Bengani H et al: Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum Mol Genet 2014; 23: 2569–2579.
Article CAS Google Scholar - Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Article CAS Google Scholar - Chomczynski P, Sacchi N : Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156–159.
Article CAS Google Scholar - Zhi J, Hatchwell E : Human MLPA Probe Design (H-MAPD): a probe design tool for both electrophoresis-based and bead-coupled human multiplex ligation-dependent probe amplification assays. BMC Genomics 2008; 9: 407.
Article Google Scholar - Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G : Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Article Google Scholar
Acknowledgements
We thank the girl’s parents for their consent and the possibility to publish this case. This study is part of a project that is the winner of the Trinational Metropolitan Region’s ‘Offensive Sciences’ and co-financed by the European Union’s European Fund for Regional Development in the framework of the ‘INTERREG IV Upper Rhine Program’.
Author information
Author notes
- Ann-Sophie Kaiser and Bianca Maas: These authors contributed equally to this work.
Authors and Affiliations
- Institute of Human Genetics, Heidelberg University, Heidelberg, Germany
Ann-Sophie Kaiser, Bianca Maas, Christian Sutter, Johannes WG Janssen, Katrin Hinderhofer & Ute Moog - Department of Conservative Dentistry, Heidelberg University, Heidelberg, Germany
Anna Wolff
Authors
- Ann-Sophie Kaiser
You can also search for this author inPubMed Google Scholar - Bianca Maas
You can also search for this author inPubMed Google Scholar - Anna Wolff
You can also search for this author inPubMed Google Scholar - Christian Sutter
You can also search for this author inPubMed Google Scholar - Johannes WG Janssen
You can also search for this author inPubMed Google Scholar - Katrin Hinderhofer
You can also search for this author inPubMed Google Scholar - Ute Moog
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toAnn-Sophie Kaiser.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kaiser, AS., Maas, B., Wolff, A. et al. Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia.Eur J Hum Genet 23, 704–707 (2015). https://doi.org/10.1038/ejhg.2014.163
- Received: 05 March 2014
- Revised: 03 July 2014
- Accepted: 09 July 2014
- Published: 13 August 2014
- Issue Date: May 2015
- DOI: https://doi.org/10.1038/ejhg.2014.163