Regulation and function of innate and adaptive interleukin‐17‐producing cells (original) (raw)
IL‐17‐mediated immunity is key for mucosal host defence against a range of pathogens, but can also elicit or aggravate autoimmune and cardiovascular disease, as well as affect tumour formation. The regulation and function of such versatile cells is discussed here.
Introduction
Interleukin‐17 (IL‐17) is a proinflammatory cytokine secreted by innate and adaptive immune cells (Cua & Tato, 2010; Hirota et al, 2010; Korn et al, 2009). Although the source of IL‐17 is restricted to haematopoietic cells, the heterodimeric IL‐17 receptor—consisting of IL‐17 RA and IL‐17 RC—is widely expressed by haematopoietic and non‐haematopoietic cells, such as endothelial and epithelial cells. IL‐17 downstream signalling is regulated by the complex IL‐17R–Act1–TRAF6, which is antagonized by TRAF3 (Zhu et al, 2010). Therefore, potential target cell types, tissues and organs are also variable, depending on when and where an IL‐17‐mediated immune response occurs.
There are several types of haematopoietic cell known to produce IL‐17, including αβT, γδT and innate lymphocytes negative for lineage markers (Cupedo et al, 2009; Takatori et al, 2009). The predominant source of IL‐17 depends on the inflammatory and physiological microenvironment, which provides the condition for a particular cell type to secrete IL‐17. Analysis of an IL‐17 Cre fate reporter mouse emphasized that, under steady‐state conditions, most of the IL‐17‐producing cells are CD4 T cells found in the lamina propria of the small intestine, which also contains a population of innate lymphocytes, as previously determined with intracellular staining approaches (Hirota et al, 2011). Furthermore, there are γδ T cells in the skin (dermal γδ T cells) that express IL‐17 (Gray et al, 2011), as well as a substantial population of a γδ T‐cell subset that expresses Vγ4 and IL‐17 in the lung (Fig 1; J.H.D. & B.S., unpublished observations).
Figure 1
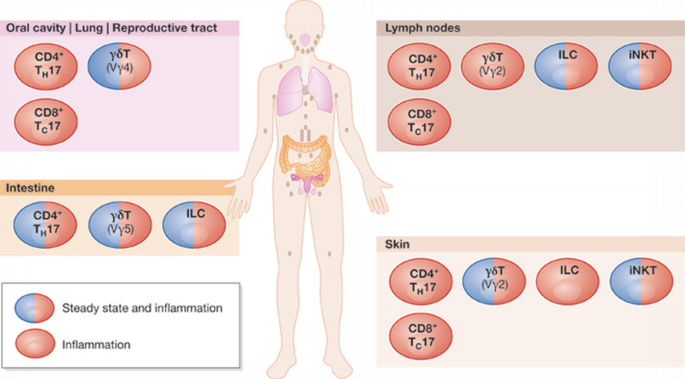
Location of IL‐17‐producing cell types. There are characteristic populations of homeostatic IL‐17‐producing cell types in different mucosal tissues. Upon inflammation, these cells act in their resident tissue or migrate to other tissues to combat infection. For example, LN CD4+ TH17 cells can give rise to inflammatory CD4+ TH17 cells that migrate to many tissues in the body. ILC, innate lymphoid cell; iNKT, invariant natural killer T cell; LN, lymph node.
In line with the clear phenotypes of IL‐17‐deficient mice in various disease models, high levels of IL‐17 are induced in response to a range of infections, as well as during the development of several autoimmune diseases.
To understand IL‐17‐mediated immune responses, several facets of this response have to be taken into account, as the IL‐17 receptor is ubiquitously expressed throughout the body, resulting in pleiotropic actions of IL‐17 on a wide range of cell types. One of the well‐known biological outcomes of IL‐17 signalling is the induction of proinflammatory cytokines and chemokines from target cells, such as fibroblasts and epithelial cells, leading to the recruitment of neutrophils (Fossiez et al, 1998). The primary function of activated neutrophils is to fight against invading pathogens, but this can damage the surrounding tissues.
Although the mechanisms that underlie the innate IL‐17 response remain largely unknown, the differentiation, maintenance and effector function of IL‐17‐producing CD4 T cells have been extensively studied. Naive CD4 T helper cells can differentiate into IL‐17‐producing T cells, known as TH17 cells. This particular subset of T helper cells can secrete not only the signature cytokine IL‐17, but also TNF‐α, GM‐CSF, IL‐2, IL‐17F and IL‐22, depending on inflammatory settings. IL‐23 and environmental signals play a crucial role in the pathogenesis of TH17 cells in autoimmune settings (Stockinger et al, 2011).
Here, we review the latest discoveries regarding the regulation and function of innate and adaptive IL‐17‐producing cells in the context of beneficial and adverse immune responses.
Transcriptional regulation of IL‐17‐positive cells
There have recently been major advances in our understanding of the transcriptional regulation of TH17 cells. Although both innate and adaptive IL‐17‐producing cells express the master transcription factor Rorγt, the differentiation and regulatory factors seem to differ between innate and adaptive cells. Most studies show a requirement for IL‐6 signalling in TH17 development, but notably IL‐6 is irrelevant for the development and maintenance of IL‐17‐producing γδ T cells as well as iNKT cells (Doisne et al, 2009; Martin et al, 2009). How the differentiation of IL‐17‐producing innate lymphocytes is regulated is unknown (Sidebar A). Several papers have shown that TGF‐β, in combination with IL‐6, is required for the induction of Rorγt and TH17 development in mice and, in addition, the IL‐21 and IL‐23 signalling pathways amplify TH17 differentiation (Hirota et al, 2010; Korn et al, 2009). In line with this, the development of IL‐17‐producing γδ T cells is severely impaired in TGF‐β‐deficient mice, suggesting a crucial role of TGF‐β signalling (Do et al, 2010). Nevertheless, this idea was challenged in a study suggesting that TGF‐β‐independent differentiation of TH17 cells occurs through IL‐23 in combination with IL‐6 and IL‐1 (Ghoreschi et al, 2010). However, it must be noted that, despite the use of neutralizing antibodies against TGF‐β, it might be impossible to remove all sources of TGF‐β, as a cell‐associated form of TGF‐β—produced by activated T cells—cannot be neutralized by the widely used TGF‐β antibody (Clone:1D11; Oida & Weiner, 2011). Thus, it cannot be excluded that TGF‐β generated in activated T cells and acting on bystander cells might have a role in TH17 differentiation. Although TGF‐β was initially reported in human studies to be redundant in generating TH17 cells (Acosta‐Rodriguez et al, 2007; Wilson et al, 2007), other groups demonstrated subsequently that TGF‐β plays a crucial role in the induction of Rorγt and differentiation of human TH17 (Manel et al, 2008; O'Garra et al, 2008; Volpe et al, 2008). However, it has been suggested that TGF‐β is required only to block the alternative T helper cell differentiation pathways, instead of directly affecting TH17 differentiation (Das et al, 2009). Indeed, TGF‐β signalling suppresses the induction of TH17 inhibitors such as SOCS3, a well‐known negative regulator of STAT3, and blocks the induction of eomesodermin in a Smad‐independent manner, indirectly promoting TH17 differentiation (Ichiyama et al, 2011; Qin et al, 2009). Furthermore, the role of Smad2 and Smad3 signalling downstream from TGF‐β during TH17 development is not without controversy, as one study shows that both Smad2 and Smad3 are necessary for the optimal induction of TH17 cells (Takimoto et al, 2010), whereas another indicates that Smad3 deficiency increases TH17 differentiation (Martinez et al, 2009). Although there still remain unresolved issues, it is conceivable that TGF‐β promotes TH17 differentiation directly as well as indirectly.
Sidebar A | In need of answers
- i.
Which factors regulate the differentiation of IL‐17‐producing innate lymphoid cells? Where are they induced? - ii.
Can Rorγt antagonists limit IL‐17‐mediated tissue pathology in human diseases? - iii.
Which factors and conditions drive TH17 plasticity into different cytokine profiles? - iv.
Can ‘ex‐TH17’ cells revert to their original state? - v.
Does TH17 memory—in the absence of any triggering stimulus—exist?
A substantial amount of work has concentrated on the signalling and transcriptional events that occur during TH17 development, influencing the induction and maintenance of their effector profile (Fig 2). The transcription factors Rorγt, AhR and IκBζ are specifically induced during TH17‐cell development and have key roles in their differentiation and function (Okamoto et al, 2010; Veldhoen et al, 2008). STAT3, IRF4, Runx1 and Batf, on the other hand, influence TH17 development, but their expression is not restricted to TH17 cells (Korn et al, 2009; Schraml et al, 2009). As anticipated, the transcriptional modulators that target these key transcription factors can either enhance or impair the differentiation of TH17 cells. The positive and negative regulators of the key transcription factor IRF4—ROCK2 and IRF4 binding protein, respectively—were clearly shown to alter the activity of IRF4, thereby resulting in the spontaneous development of autoimmune disorders in mice with aberrantly activated ROCK2 and deficient in IRF4 binding protein (Biswas et al, 2010; Chen et al, 2008). The nuclear receptor LXR negatively regulates the IL‐17 immune response by upregulating the transcription factor Srebp‐1c, which interacts physically with AhR to interfere with its function in promoting TH17 development (Cui et al, 2011). In addition, the nuclear receptor PPARγ was shown to negatively regulate TH17 differentiation by repressing the induction of Rorγt. However, the precise mechanisms by which PPARγ interferes with STAT3, IRF4 and Batf‐mediated Rorγt induction remain to be elucidated (Klotz et al, 2009). The transcription factor TCF1 inhibits transcription of IL‐17 in a different manner, by directly binding to the regulatory region of the IL‐17 gene (Yu et al, 2011).
Figure 2
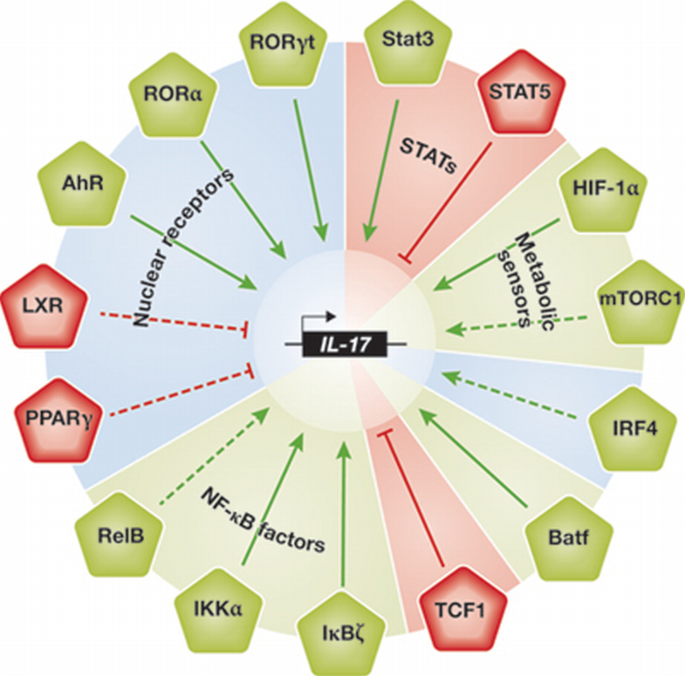
Transcriptional regulation of the IL‐17 programme. Members of various transcription factor families are implicated in the regulation of IL‐17 transcription either directly (solid line) or indirectly (dashed line). Green indicates positive regulation and red negative regulation. AhR, aryl hydrocarbon receptor; Batf, basic leucine zipper transcription factor, ATF‐like; HIF, hypoxia‐inducible factor; IκB, inhibitor of NF‐κB; IKK, inhibitor of NF‐κB kinase; IRF, interferon response factor; LXR, liver X receptor; mTORC, mammalian target of rapamycin complex; NF‐κB, nuclear factor κB; PPAR, peroxisome proliferator‐activated receptor; RelB, v‐rel reticuloendotheliosis viral oncogene homologue B; ROR, retinoid‐related orphan nuclear receptor; STAT, signal transducer and activator of transcription; TCF, T‐cell‐specific transcription factor.
Similarly to the differential role of STAT4 and STAT6 for TH1 and TH2 differentiation, respectively, STAT3 downstream from IL‐6 signalling has a central role in the initiation of TH17 development. The negative effect of IL‐2 signalling on TH17 differentiation has been shown recently to be mediated by STAT5 activity. This activity competes with STAT3 for binding to multiple common sites of the IL‐17 promoter region and directly antagonizes IL‐17 transcription in part through an epigenetic modification (Yang et al, 2011).
The components of the NF‐κB signalling pathway regulate numerous immune responses, and their dysregulation is linked to inflammatory and autoimmune diseases, as well as cancer (Vallabhapurapu & Karin, 2009). Interestingly, several of these molecules regulate Rorγt and IL‐17A expression. Thymic γδT cells express the surface lymphotoxin‐β‐receptor and, after its ligation, RelB mediates the induction of Rorγt in γδ T cells, but not αβ T cells, which is required for the differentiation of IL‐17‐producing γδ T cells and an innate IL‐17 response to Escherichia coli infection (Powolny‐Budnicka et al, 2011). This unique contribution of RelB to innate IL‐17‐producing cells suggests the different functional regulation of innate and adaptive IL‐17 production. Besides NF‐κB, kinase inhibitors for NF‐κB signalling also play a role in TH17 differentiation. Although IKKα was originally identified as an inhibitor of NF‐κB, IKKα can promote IL‐17 transcription in TH17 cells by binding to the IL‐17 locus in a NF‐κB‐independent way (Li et al, 2011). Furthermore, IκBζ is specifically induced during TH17 differentiation and, in cooperation with Rorγt, is essential for robust IL‐17 transcription (Okamoto et al, 2010).
Several groups have demonstrated recently that the kinase mTOR and the transcription factor HIF1‐α, which are cellular metabolic sensors, control TH17 fate determination. The mTOR complex 1 (mTORC1), but not mTORC2, regulates the small GTPase Rheb signalling pathway, which promotes TH1 and TH17, but not TH2, differentiation (Delgoffe et al, 2011). In addition, the detailed mechanisms by which HIF‐1α affects TH17, but not TH1, cell fate have been defined. HIF‐1α is upregulated during TH17 differentiation, possibly in a STAT3‐ and mTOR‐dependent manner, which in turn enhances Rorγt expression. After this event, a molecular complex with STAT3, HIF‐1α, Rorγt and P300 induces the robust expression of TH17‐associated genes (Dang et al, 2011; Shi et al, 2011).
Mucosal host defence
IL‐17 is a potent proinflammatory cytokine that sets in motion the recruitment of neutrophils and monocytes in tissues and induces antimicrobial peptide production by epithelial barrier cells. In animal models of infection, IL‐17‐deficient mice are highly susceptible to bacterial and fungal infections. Coinciding with the discovery of IL‐17 function in mouse models, two independent groups identified a crucial role for IL‐17 in the defence against fungal and bacterial infections in humans (Fig 3). Dominant‐negative mutations in human STAT3 lead to the development of hyper‐IgE syndrome, which compromises the generation of IL‐17‐producing cells. These patients suffer from repeated mucocutaneous candidiasis (CMCD) and pulmonary infections with Staphylococcus aureus (Holland et al, 2007; Minegishi et al, 2007). STAT3 loss‐of‐function reduces the number of IL‐17‐producing cells, the differentiation of which in vitro is impaired due to the essential role of STAT3 downstream from IL‐6 in the development of TH17 (de Beaucoudrey et al, 2008; Ma et al, 2008; Milner et al, 2008; Minegishi et al, 2009). In addition, IL‐17F or IL‐17 receptor A deficiency was linked to Candida infection (Puel et al, 2011). In other cases of CMCD, IL‐17‐producing cells were competent, but the patients generated neutralizing antibodies to IL‐17 (Kisand et al, 2010). Furthermore, autosomal‐recessive mutation of CARD9 confers susceptibility to Candida infection (Glocker et al, 2009). CARD9 is an adaptor molecule that, in macrophages and dendritic cells, acts downstream from the pattern recognition receptors dectin 1 and dectin 2, which are activated by β‐glucan—a component of the fungal cell wall—and drive IL‐17 immunity (LeibundGut‐Landmann et al, 2007; Robinson et al, 2009; Saijo et al, 2010). Finally, gain‐of‐function mutations in STAT1, which strongly inhibit the development of IL‐17‐producing cells, were found in patients with CMCD (Liu et al, 2011). Despite the fact that the phenotype of these genetic mutants clearly established the importance of IL‐17 for defence against Candida infection, the cellular origin of the cytokine had not been identified. IL‐17‐producing γδ T cells, but not αβ T cells, have an important role in skin S. aureus infection (Cho et al, 2010). Thus, this γδ T‐cell subset acts as a first line of defence in the IL‐17‐mediated immune response and orchestrates the following inflammatory TH17 response. To efficiently sense invading pathogens, IL‐17‐producing γδ T cells highly express the Toll‐like receptors TLR1 and TLR2, as well as dectin 1. Signalling from these molecules, together with environmental stimuli, regulate the magnitude of the IL‐17 immune response and subsequent neutrophil activation (Martin et al, 2009).
Figure 3
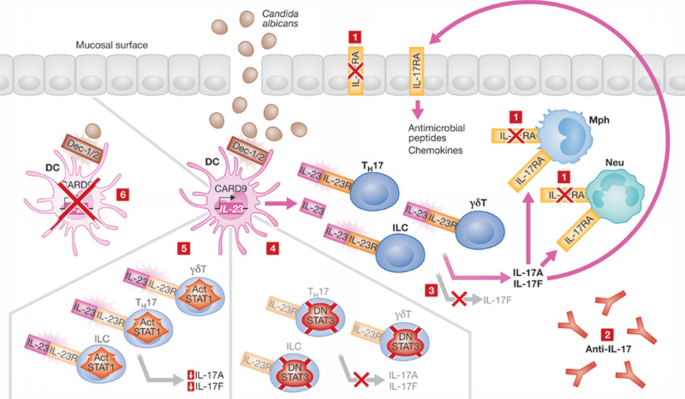
Defects and mutations resulting in mucocutaneous candidiasis. Candida albicans is recognized by dectin 1/2 in dendritic cells, which induces the CARD9‐mediated transcription of IL‐23. IL‐23 is recognized by IL‐23R, which is expressed by TH17, innate lymphocyte and γδT cells. This induces IL‐17A and IL‐17F expression, which modulate the immune response by inducing the expression of antimicrobial peptides and chemokines through binding to IL‐17RA. TH17 differentiation and IL‐17 cytokine expression is positively regulated by STAT3, whereas STAT1 activation antagonizes TH17 cell differentiation. Mucocutaneous candidiasis is caused by various defects in this immune response pathway, including (i) IL‐17RA deficiency, (ii) autoantibodies for IL‐17, (iii) lack of expression of IL‐17F, (iv) mutations resulting in the dominant‐negative form of STAT3, (v) expression of a hyperactivated (Act) form of STAT1, or (vi) CARD9 deficiency. CARD, caspase recruitment domain family; DC, dendritic cell; Dec, dectin; IL, interleukin; ILC, innate lymphocyte; STAT, signal transducer and activator of transcription; TH, T helper.
The elucidation of the environmental signals that control the development and maintenance of mucosal TH17 cells in the intestine remained an intriguing question. Recent studies have revealed that colonization of the gut with TH17 cells requires distinct commensal bacteria, such as segmented filamentous bacteria (Gaboriau‐Routhiau et al, 2009; Ivanov et al, 2009). TH17 colonization of the gut increases the level of antimicrobial defences and confers resistance to intestinal pathogens, such as Citrobacter and Salmonella, in a NOD1/NOD2‐dependent manner (Geddes et al, 2011). The colonization of the intestine with TH17 cells also has an impact on systemic immunity. In fact, germ‐free conditions protected mice against the development of autoimmune arthritis in a model dependent on IL‐17 production and germinal centre formation for autoantibody‐secreting B cells. The introduction of segmented filamentous bacteria into such germ‐free mice restored the intestinal TH17 population and B cells causing autoantibody‐mediated arthritis (Wu et al, 2010). Thus, the induction of TH17 cells in the intestinal lamina propria might contribute not only to mucosal host defence, but also to the induction of autoimmunity in other tissues. Interestingly, intestinal TH17 cells are entirely dependent on commensal flora, whereas germ‐free mice contain numbers of IL‐17‐producing γδ T cells in the skin and lung comparable with those in mice under specific pathogen‐free conditions (J.H.D. & B.S., unpublished data). These findings suggest that, in addition to the distinct requirement of cytokines between innate and adaptive IL‐17‐producing cells, environmental factors strikingly affect their distribution and function.
Adverse effects of IL‐17‐mediated immune responses
In contrast to the beneficial effect of IL‐17 on host defence, IL‐17‐mediated immune responses can have adverse effects resulting in tissue damage. Secondary tissue damage often occurs after infection. For example, although IL‐17 signalling is not required for the function of cytotoxic CD8 T cells and viral clearance after flu infection, bystander IL‐17‐mediated neutrophil influx can cause lung damage (Crowe et al, 2009). Besides the infection‐related pathology, unexpected adverse effects linked to IL‐17 were discovered in cardiovascular diseases, ischaemic brain injury and tumorigenesis, which also involve immune cells and microinflammation. Both TH1 and TH17 cells infiltrate atherosclerotic plaques in human coronary arteries, and the combination of IFN‐γ and IL‐17 induces proinflammatory cytokines and chemokines that might recruit pathogenic monocytes and macrophages to atherosclerotic lesions (Eid et al, 2009). By using apoE‐deficient mice—an animal model of atherosclerosis—IL‐17, produced mainly by TH17 and γδ T cells, was shown to have a crucial role in the induction of IL‐6, CCL5 and CXCL1 from aortic endothelial cells. This induction recruits other inflammatory cells and enlarges atherosclerotic lesions (Erbel et al, 2009; Smith et al, 2010). Furthermore, an upregulation of IL‐17 was detected in human stable atherosclerotic lesions (Taleb et al, 2009). On the other hand, IL‐17 seems to induce an immunosuppressive macrophage phenotype, which limits vascular inflammation and atherosclerosis lesion development. In addition to cardiovascular diseases, sterile inflammation is also observed due to danger signals released from necrotic cells after ischaemic brain injury. The recruitment and activation of immune cells sensing danger signals clearly cause secondary injury in the late phase. Activated γδ T cells are a main source of IL‐17 and a pathogenic effector in the late phase of ischaemic injury after the activation of infiltrating macrophages and production of IL‐23 (Shichita et al, 2009).
Another type of microinflammation is tumour‐associated immunity. Immune surveillance mediated by NK, TH1 and cytotoxic CD8 T cells is essential to prevent and inhibit tumour formation. Nonetheless, chronic inflammation is often related to the initiation of tumour formation. The enterotoxigenic _Bacteroides fragilis_—a common human commensal bacterium—induces chronic IL‐17‐mediated colonic inflammation through which colonic hyperplasia and carcinogenesis are initiated in a multiple intestinal neoplasia mouse model (Wu et al, 2009). IL‐17 has also been associated with the formation of giant cells in Langerhans cell histiocytosis, which establishes dendritic cells as important targets for IL‐17 in this disease (Coury et al, 2008).
Innate and adaptive IL‐17 responses enable biological barriers against pathogens. Although innate IL‐17 responses might suffice to eradicate most pathogens, excess innate immune reactions might lead to hyperinflammatory responses, resulting for example in allergy in the lung and inflammatory diseases. Adaptive IL‐17 cells constitute the second stage of the immune reaction and might be recruited when the innate response fails to cope with invading pathogens. Pathogen‐specific TH17 cells raise a strong coordinated immune response, whereas IL‐17‐mediated autoimmune disorders can be initiated by autoreactive TH17 cells, after tissue injury induces the release of self‐antigen. Autoimmune diseases that are linked to IL‐17 are invariably dependent on adaptive T cells, whereas innate IL‐17 producers are either not involved or have merely accessory roles.
Several diseases that are considered to be of autoimmune origin are clearly associated with TH17 cells, and deficiencies in TH17 cytokines or transcription factors regulating the development and function of TH17 cells often reduce pathology in various disease models (Hirota et al, 2010; Korn et al, 2009). Genome‐wide association studies demonstrated that the TH17‐related molecules IL‐23 and chemokine receptor CCR6 are associated with inflammatory bowel disease, rheumatoid arthritis and psoriasis in humans (Duerr et al, 2006; Kochi et al, 2010; Nair et al, 2009). Nevertheless, the mechanisms that underlie pathogenicity against self are largely unknown. The primary immune response elicited by TH17 cells is associated with preferential neutrophil infiltration, and innate immune cells activated by IL‐17 could damage the surrounding tissue. Indeed, TH17 cells directly cause the damage of axons, leading to neuronal dysfunction in a contact‐dependent manner (Siffrin et al, 2010). Recent reports have revealed that not only TH17 cells, but also IL‐17‐producing innate lymphocytes significantly contribute to the pathogenicity of autoimmune diseases. In experimental autoimmune encephalitis (EAE), γδ T cells produce IL‐17 early in response to IL‐1β and IL‐23, which are secreted by macrophages and dendritic cells before the ensuing TH17 response and amplified TH17‐mediated autoimmunity (Sutton et al, 2009). Although not autoimmune in nature, other pathological states can be induced by, for example, IL‐17‐producing NKT cells in the lung. These cells initiate ozone‐induced asthma, which is characterized by airway hypersensitivity and airway neutrophilia, but not eosinophilia (Pichavant et al, 2008). Furthermore, pulmonary IL‐17‐producing γδ T cells and TH17 cells promote lung fibrosis through increased synthesis and secretion of collagen with neutrophilia in bleomycin‐exposed mice (Mi et al, 2011; Wilson et al, 2010). In contrast to the cooperative action of innate cells with TH17 cells, colonic‐lineage negative innate lymphocytes that express Rorγt and IL‐23R produce IL‐17 and IFN‐γ, causing intestinal pathology in RAG‐deficient mice independently of innate and adaptive T cells (Buonocore et al, 2010). IL‐23 seems to be an essential cytokine to drive the pathogenicity of both innate and adaptive IL‐17‐producing cells, and an upstream mechanism underlying the induction of IL‐1β, IL‐6 and IL‐23 by macrophages and dendritic cells was reported. In synergy with pattern recognition receptor signalling, the activation of serum complement pathways stimulates macrophages and dendritic cells to help initiate TH17 differentiation and activation (Hashimoto et al, 2010; Lajoie et al, 2010).
Considering the adverse effects of dysregulated IL‐17, controlling innate and adaptive IL‐17‐producing cells could be a key therapeutic target for the prevention and treatment of IL‐17‐associated tissue inflammation. The master transcription factor Rorγt universally marks innate as well as adaptive IL‐17‐producing cells. Therefore, the inhibition of Rorγt by small molecules might limit the sustainment of IL‐17 immune reactions and the ensuing tissue damage. Three independent groups have shown that the natural cardiac glycoside digoxin and high‐affinity synthetic ligands for Rorα and Rorγt inhibit the activity of these master regulators of IL‐17 transcription and block the differentiation and effector function of TH17 cells (Fujita‐Sato et al, 2011; Huh et al, 2011; Solt et al, 2011). As the safety of digoxin is already established, it could be tested for its effect on the limitation of innate and adaptive IL‐17‐mediated tissue pathology.
Plasticity of TH17 cells
Terminally differentiated effector CD4 T cells were thought to be stable, but numerous recent studies have identified factors that reinforce or destabilize effector‐T‐cell programmes (Murphy & Stockinger, 2010). TH17 cells in particular seem to disobey the paradigm of irreversible commitment enunciated for TH1 and TH2 differentiation. Although ex vivo isolated TH17 cells appear relatively stable, _in vitro_‐generated TH17 cells are substantially plastic and can turn into IFN‐γ‐producing, TH1‐like, or IL‐4‐producing, TH2‐like cells in response to IL‐12 or IL‐4, respectively (Lexberg et al, 2008). Furthermore, _in vitro_‐generated TH17 cells can cause IFN‐γ‐dependent type 1 diabetes and colitis after transfer into lymphopaenic recipient mice (Bending et al, 2009; Lee et al, 2009; Nurieva et al, 2009). Epigenetic analysis revealed that _in vitro_‐generated TH17 cells have DNA modifications accessible to TH1 and TH2 lineage transcription factors, whereas _in vitro_‐generated TH1 and TH2 cells have epigenetic marks rendering IL‐17A and Rorγt inaccessible (Mukasa et al, 2010). To investigate the plasticity of TH17 cells in vivo, we generated a reporter mouse strain designed to fate‐map cells that have activated IL‐17. IL‐17 fate reporter mice show that TH17 cells developing during chronic inflammatory responses in the EAE model rapidly shut down expression of IL‐17A and switch on alternative cytokines, whereas in a model of acute cutaneous Candida infection, TH17 cells do not deviate to alternative cytokine production. During the development of EAE, TH17, rather than TH1, cells are the main source of IFN‐γ and other pro‐inflammatory cytokines in the spinal cord. Of note, T‐bet+ IFN‐γ‐producing ‘ex‐TH17’ cells driven by IL‐23 retain specific markers of the TH17 programme (Hirota et al, 2011). IL‐23 was also shown to differentiate T cells into a pathogenic IL‐17‐ and IFN‐γ‐producing subset in a colitis model (Ahern et al, 2010). Furthermore, T‐bet‐deficient T cells fail to develop into TH1‐like cells from ex‐TH17 cells (Lazarevic et al, 2011). Our data suggest that the vast majority of proinflammatory cytokine‐producing cells in EAE are originally TH17, suggesting that encephalitogenic T cells in EAE are primarily ex‐TH17 cells. This would change the interpretation of work reported by two independent groups showing that GM‐CSF‐producing T cells—instead of IFN‐γ‐producing or IL‐17‐producing T cells—are a pathogenic subset in EAE (Codarri et al, 2011; El‐Behi et al, 2011). Intriguingly, it has been shown that, in human arthritis, a large population of synovial IL‐17‐negative IFN‐γ‐producing TH1 cells express RORC2, CCR6 and CD161, which are markers of human TH17 cells (Nistala et al, 2010). In contrast to the deviation of TH17 cells to alternative proinflammatory cytokines in EAE, ex‐TH17 cells are not converted to a regulatory‐T‐cell phenotype that expresses FoxP3 or IL‐10 in this setting, neither in the draining lymph node nor in the spinal cord. However, in a model of tolerance induced by CD3‐specific antibody, activated CCR6+ TH17 cells are intriguingly redirected to the small intestine by chemotaxis to CCL20, which is secreted by the intestinal epithelial cells. Once there, the proinflammatory TH17 cells acquire a regulatory phenotype with in vitro and in vivo immune‐suppressive properties (Esplugues et al, 2011).
Memory is a central feature of the adaptive immune system. Given that TH17 cells easily switch off their signature cytokine and acquire alternative profiles in some immune responses, it is difficult to trace adaptive IL‐17 producers through the assessment of intracellular cytokine profiles. Indeed, based on cytokine staining, it has been suggested that there are no memory phenotype cells expressing IL‐17 (Pepper et al, 2010). The fate reporter system should allow a more detailed assessment of this issue. In agreement with our fate reporter study showing that a proportion of eYFP+ TH17 cells lacked IL‐17 expression ex vivo even after PdbU/ion restimulation, a recent study in human subjects also suggested that a proportion of human CCR6+ memory TH17 cells do not produce IL‐17 ex vivo after restimulation. Intriguingly, recall responses resulting in IL‐17 production were induced from IL‐17 CCR6+, but not CCR6− cells after in vitro culture with common γ‐chain cytokines, such as IL‐2, IL‐7 and IL‐15 (Wan et al, 2011). Further studies of memory formation and recall of distinct effector profiles are needed to understand the complexities of recalling memory cells into effector responses tailored to combat different types of pathogen that require specific cytokine responses (Sidebar A).
Concluding remarks
Accumulating evidence suggests that inborn errors of IL‐17‐mediated immune pathways precipitate CMCD and other infections in affected patients, emphasizing the crucial role of this cytokine for host defence against a range of pathogens. On the other hand, genome‐wide association studies have identified TH17‐related genes as contributors or initiators of autoimmune diseases such as multiple sclerosis, rheumatoid arthritis and psoriasis. These results clearly suggest that the activation and maintenance of IL‐17‐producing cells should be robust during infection and at the same time tightly regulated to avoid the risk of autoinflammatory disorders. Therefore, understanding the multiple steps of the IL‐17‐mediated immune response would help to develop future immunotherapy approaches for both infectious and autoimmune diseases. As the regulators of differentiation, maintenance and effector function between innate and adaptive IL‐17‐producing cells seem to be different, it is important to understand the distinct role and effector regulation of innate and adaptive IL‐17‐producing cell types in a variety of immune conditions. Lastly, epigenetic DNA modification and microRNAs are important factors in gene regulation, which can substantially modify the levels of target gene expression. Our understanding of IL‐17 regulation by epigenetic mechanisms and microRNAs at a mechanistic level in vivo is still incomplete. Future research and integrated information will hopefully define improved targeted therapies based on transcriptional and post‐transcriptional IL‐17 gene regulation and an effective control of beneficial and adverse IL‐17‐mediated immune responses.
Act1:
actin‐related gene 1
AhR:
aryl hydrocarbon receptor
apoE:
apolipoprotein E
Batf:
basic leucine zipper transcription factor, ATF‐like
CARD:
caspase recruitment domain family
CCL:
chemokine (C‐C motif) ligand
CCR:
chemokine (C‐C motif) receptor
CXCL:
chemokine (C‐X‐C motif) ligand
CD161:
killer cell lectin‐like receptor subfamily B member 1C
eYFP:
enhanced yellow fluorescent protein
FoxP3:
forkhead box P3
GM‐CSF:
granulocyte monocyte colony stimulating factor
HIF‐1α:
hypoxia‐inducible factor 1a
IFN‐γ:
interferon gamma
IκBζ:
inhibitor of κBζ
IL:
interleukin
IRF4:
interferon regulatory factor 4
iNKT cell:
invariant natural killer T cell
IKK‐α:
inhibitor of NF‐κB kinase alpha
LXR:
liver X receptor
mTOR:
mammalian target of rapamycin
NF‐κB:
nuclear factor κB
NK:
natural killer
NKT:
natural killer T
NOD:
nucleotide‐binding oligomerization domain
PdbU/ion:
phorbol 12,13‐dibutyrate/ionomycin
PPARγ:
peroxisome proliferator‐activated receptor gamma
RAG:
recombination activating gene
RelB:
reticuloendotheliosis viral (v‐rel) oncogene related B
Rheb:
Ras homologue enriched in brain
ROCK2:
Rho‐associated protein kinase 2
RORC2:
RAR‐related orphan receptor C2
Rorγt:
RAR‐related orphan receptor gamma t
Runx1:
runt‐related transcription factor 1
SOCS3:
suppressor of cytokine signalling 3
Srebp‐1c:
sterol regulatory element binding transcription factor 1c
STAT:
signal transducer and activator of transcription
T‐bet:
T‐box 21
TGF‐β:
transforming growth factor beta
TH:
T helper
TNF:
tumour necrosis factor
TRAF:
TNF‐receptor‐associated factor
References
- Acosta‐Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1beta and 6 but not transforming growth factor‐beta are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat Immunol 8: 942–949
Google Scholar - Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F (2010) Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity 33: 279–288
Google Scholar - Bending D, De la Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A (2009) Highly purified Th17 cells from BDC2.5NOD mice convert into Th1‐like cells in NOD/SCID recipient mice. J Clin Invest 119: 565–572
Google Scholar - Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, Pernis AB (2010) Phosphorylation of IRF4 by ROCK2 regulates IL‐17 and IL‐21 production and the development of autoimmunity in mice. J Clin Invest 120: 3280–3295
Google Scholar - Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin‐23‐dependent innate intestinal pathology. Nature 464: 1371–1375
Google Scholar - Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, Pernis AB (2008) IRF‐4‐binding protein inhibits interleukin‐17 and interleukin‐21 production by controlling the activity of IRF‐4 transcription factor. Immunity 29: 899–911
Google Scholar - Cho JS et al (2010) IL‐17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest 120: 1762–1773
Google Scholar - Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) RORgammat drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560–567
Google Scholar - Coury F et al (2008) Langerhans cell histiocytosis reveals a new IL‐17A‐dependent pathway of dendritic cell fusion. Nat Med 14: 81–87
Google Scholar - Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK (2009) Critical role of IL‐17RA in immunopathology of influenza infection. J Immunol 183: 5301–5310
Google Scholar - Cua DJ, Tato CM (2010) Innate IL‐17‐producing cells: the sentinels of the immune system. Nat Rev Immunol 10: 479–489
Google Scholar - Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, Sheng H, Xi B, Zhang JZ, Zang YQ (2011) Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest 121: 658–670
Google Scholar - Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H (2009) Human fetal lymphoid tissue‐inducer cells are interleukin 17‐producing precursors to RORC+ CD127+ natural killer‐like cells. Nat Immunol 10: 66–74
Google Scholar - Dang EV et al (2011) Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell 146: 772–784
Google Scholar - Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, Van Kaer L, Shi Y, Das G (2009) Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med 206: 2407–2416
Google Scholar - de Beaucoudrey L et al (2008) Mutations in STAT3 and IL12RB1 impair the development of human IL‐17‐producing T cells. J Exp Med 205: 1543–1550
Google Scholar - Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD (2011) The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12: 295–303
Google Scholar - Do JS, Fink PJ, Li L, Spolski R, Robinson J, Leonard WJ, Letterio JJ, Min B (2010) Cutting edge: spontaneous development of IL‐17‐producing gamma delta T cells in the thymus occurs via a TGF‐beta 1‐dependent mechanism. J Immunol 184: 1675–1679
Google Scholar - Doisne JM, Becourt C, Amniai L, Duarte N, Le Luduec JB, Eberl G, Benlagha K (2009) Skin and peripheral lymph node invariant NKT cells are mainly retinoic acid receptor‐related orphan receptor (gamma)t+ and respond preferentially under inflammatory conditions. J Immunol 183: 2142–2149
Google Scholar - Duerr RH et al (2006) A genome‐wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314: 1461–1463
Google Scholar - Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, Sokol SI, Pfau S, Pober JS, Tellides G (2009) Interleukin‐17 and interferon‐gamma are produced concomitantly by human coronary artery‐infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation 119: 1424–1432
Google Scholar - El‐Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol 12: 568–575
Google Scholar - Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, Wang Y, Bockler D, Katus HA, Dengler TJ (2009) Inhibition of IL‐17A attenuates atherosclerotic lesion development in apoE‐deficient mice. J Immunol 183: 8167–8175
Google Scholar - Esplugues E et al (2011) Control of TH17 cells occurs in the small intestine. Nature 475: 514–518
Google Scholar - Fossiez F, Banchereau J, Murray R, Van Kooten C, Garrone P, Lebecque S (1998) Interleukin‐17. Int Rev Immunol 16: 541–551
Google Scholar - Fujita‐Sato S, Ito S, Isobe T, Ohyama T, Wakabayashi K, Morishita K, Ando O, Isono F (2011) Structural basis of digoxin that antagonizes ROR{gamma}t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)‐17 production. J Biol Chem 286: 31409–31417
Google Scholar - Gaboriau‐Routhiau V et al (2009) The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31: 677–689
Google Scholar - Geddes K et al (2011) Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17: 837–844
Google Scholar - Ghoreschi K et al (2010) Generation of pathogenic T(H)17 cells in the absence of TGF‐beta signalling. Nature 467: 967–971
Google Scholar - Glocker EO et al (2009) A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361: 1727–1735
Google Scholar - Gray EE, Suzuki K, Cyster JG (2011) Cutting edge: Identification of a motile IL‐17‐producing gammadelta T cell population in the dermis. J Immunol 186: 6091–6095
Google Scholar - Hashimoto M et al (2010) Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med 207: 1135–1143
Google Scholar - Hirota K, Martin B, Veldhoen M (2010) Development, regulation and functional capacities of Th17 cells. Semin Immunopathol 32: 3–16
Google Scholar - Hirota K et al (2011) Fate mapping of IL‐17‐producing T cells in inflammatory responses. Nat Immunol 12: 255–263
Google Scholar - Holland SM et al (2007) STAT3 mutations in the hyper‐IgE syndrome. N Engl J Med 357: 1608–1619
Google Scholar - Huh JR et al (2011) Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature 472: 486–490
Google Scholar - Ichiyama K et al (2011) Transcription factor Smad‐independent T helper 17 cell induction by transforming‐growth factor‐beta is mediated by suppression of eomesodermin. Immunity 34: 741–754
Google Scholar - Ivanov II et al (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498
Google Scholar - Kisand K et al (2010) Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17‐associated cytokines. J Exp Med 207: 299–308
Google Scholar - Klotz L et al (2009) The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell‐intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 206: 2079–2089
Google Scholar - Kochi Y et al (2010) A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet 42: 515–519
Google Scholar - Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL‐17 and Th17 cells. Annu Rev Immunol 27: 485–517
Google Scholar - Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, Budelsky AL, Wills‐Karp M (2010) Complement‐mediated regulation of the IL‐17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 11: 928–935
Google Scholar - Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH (2011) T‐bet represses T(H)17 differentiation by preventing Runx1‐mediated activation of the gene encoding RORgammat. Nat Immunol 12: 96–104
Google Scholar - Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT (2009) Late developmental plasticity in the T helper 17 lineage. Immunity 30: 92–107
Google Scholar - LeibundGut‐Landmann S et al (2007) Syk‐ and CARD9‐dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8: 630–638
Google Scholar - Lexberg MH, Taubner A, Forster A, Albrecht I, Richter A, Kamradt T, Radbruch A, Chang HD (2008) Th memory for interleukin‐17 expression is stable in vivo. Eur J Immunol 38: 2654–2664
Google Scholar - Li L, Ruan Q, Hilliard B, Devirgiliis J, Karin M, Chen YH (2011) Transcriptional regulation of the Th17 immune response by IKK(alpha). J Exp Med 208: 787–796
Google Scholar - Liu L et al (2011) Gain‐of‐function human STAT1 mutations impair IL‐17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208: 1635–1648
Google Scholar - Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC (2008) Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205: 1551–1557
Google Scholar - Manel N, Unutmaz D, Littman DR (2008) The differentiation of human T(H)‐17 cells requires transforming growth factor‐beta and induction of the nuclear receptor RORgammat. Nat Immunol 9: 641–649
Google Scholar - Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M (2009) Interleukin‐17‐producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31: 321–330
Google Scholar - Martinez GJ, Zhang Z, Chung Y, Reynolds JM, Lin X, Jetten AM, Feng XH, Dong C (2009) Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem 284: 35283–35286
Google Scholar - Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, Wang XX, Liu HZ, Sun W, Hu ZW (2011) Blocking IL‐17A promotes the resolution of pulmonary inflammation and fibrosis via TGF‐{beta}1‐dependent and ‐independent mechanisms. J Immunol 187: 3003–3014
Google Scholar - Milner JD et al (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper‐IgE syndrome. Nature 452: 773–776
Google Scholar - Minegishi Y et al (2007) Dominant‐negative mutations in the DNA‐binding domain of STAT3 cause hyper‐IgE syndrome. Nature 448: 1058–1062
Google Scholar - Minegishi Y et al (2009) Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper‐IgE syndrome. J Exp Med 206: 1291–1301
Google Scholar - Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, Crawford GE, Hatton RD, Weaver CT (2010) Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32: 616–627
Google Scholar - Murphy KM, Stockinger B (2010) Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol 11: 674–680
Google Scholar - Nair RP et al (2009) Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappaB pathways. Nat Genet 41: 199–204
Google Scholar - Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj‐Elliott M, Wedderburn LR (2010) Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA 107: 14751–14756
Google Scholar - Nurieva R, Yang XO, Chung Y, Dong C (2009) Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J Immunol 182: 2565–2568
Google Scholar - O'Garra A, Stockinger B, Veldhoen M (2008) Differentiation of human T(H)‐17 cells does require TGF‐beta! Nat Immunol 9: 588–590
Google Scholar - Oida T, Weiner HL (2011) Murine CD4 T cells produce a new form of TGF‐beta as measured by a newly developed TGF‐beta bioassay. PLoS ONE 6: e18365
Google Scholar - Okamoto K, Iwai Y, Oh‐Hora M, Yamamoto M, Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, Takayanagi H (2010) IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464: 1381–1385
Google Scholar - Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, Jenkins MK (2010) Different routes of bacterial infection induce long‐lived TH1 memory cells and short‐lived TH17 cells. Nat Immunol 11: 83–89
Google Scholar - Pichavant M et al (2008) Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL‐17. J Exp Med 205: 385–393
Google Scholar - Powolny‐Budnicka I, Riemann M, Tanzer S, Schmid RM, Hehlgans T, Weih F (2011) RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin‐dependent interleukin‐17 production in gammadelta T cells. Immunity 34: 364–374
Google Scholar - Puel A et al (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin‐17 immunity. Science 332: 65–68
Google Scholar - Qin H et al (2009) TGF‐beta promotes Th17 cell development through inhibition of SOCS3. J Immunol 183: 97–105
Google Scholar - Robinson MJ et al (2009) Dectin‐2 is a Syk‐coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 206: 2037–2051
Google Scholar - Saijo S et al (2010) Dectin‐2 recognition of alpha‐mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32: 681–691
Google Scholar - Schraml BU et al (2009) The AP‐1 transcription factor Batf controls T(H)17 differentiation. Nature 460: 405–409
Google Scholar - Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H (2011) HIF1alpha‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208: 1367–1376
Google Scholar - Shichita T et al (2009) Pivotal role of cerebral interleukin‐17‐producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 15: 946–950
Google Scholar - Siffrin V et al (2010) In vivo imaging of partially reversible th17 cell‐induced neuronal dysfunction in the course of encephalomyelitis. Immunity 33: 424–436
Google Scholar - Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, Galkina E (2010) Blockade of interleukin‐17A results in reduced atherosclerosis in apolipoprotein E‐deficient mice. Circulation 121: 1746–1755
Google Scholar - Solt LA et al (2011) Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472: 491–494
Google Scholar - Stockinger B, Hirota K, Duarte J, Veldhoen M (2011) External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol 23: 99–105
Google Scholar - Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341
Google Scholar - Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O'Shea JJ (2009) Lymphoid tissue inducer‐like cells are an innate source of IL‐17 and IL‐22. J Exp Med 206: 35–41
Google Scholar - Takimoto T et al (2010) Smad2 and Smad3 are redundantly essential for the TGF‐beta‐mediated regulation of regulatory T plasticity and Th1 development. J Immunol 185: 842–855
Google Scholar - Taleb S et al (2009) Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin‐17 in atherosclerosis. J Exp Med 206: 2067–2077
Google Scholar - Vallabhapurapu S, Karin M (2009) Regulation and function of NF‐kappaB transcription factors in the immune system. Annu Rev Immunol 27: 693–733
Google Scholar - Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17‐cell‐mediated autoimmunity to environmental toxins. Nature 453: 106–109
Google Scholar - Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V (2008) A critical function for transforming growth factor‐beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)‐17 responses. Nat Immunol 9: 650–657
Google Scholar - Wan Q, Kozhaya L, Elhed A, Ramesh R, Carlson TJ, Djuretic IM, Sundrud MS, Unutmaz D (2011) Cytokine signals through PI‐3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J Exp Med 208: 1875–1887
Google Scholar - Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA (2010) Bleomycin and IL‐1beta‐mediated pulmonary fibrosis is IL‐17A dependent. J Exp Med 207: 535–552
Google Scholar - Wilson NJ et al (2007) Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 8: 950–957
Google Scholar - Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D (2010) Gut‐residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32: 815–827
Google Scholar - Wu S et al (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15: 1016–1022
Google Scholar - Yang XP et al (2011) Opposing regulation of the locus encoding IL‐17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 12: 247–254
Google Scholar - Yu Q, Sharma A, Ghosh A, Sen JM (2011) T cell factor‐1 negatively regulates expression of IL‐17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J Immunol 186: 3946–3952
Google Scholar - Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, Liu Y, Zhao L, Li X, Shi Y, Qian Y (2010) Modulation of experimental autoimmune encephalomyelitis through TRAF3‐mediated suppression of interleukin 17 receptor signaling. J Exp Med 207: 2647–2662
Google Scholar