Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D (original) (raw)
C. merolae is a small (2 µm diameter) unicellular organism that inhabits sulphate-rich hot springs (pH 1.5, 45 °C) (Fig. 1). The cells of C. merolae offer unique advantages for studies of mitochondrial and plastid (chloroplast) divisions1,2,3,4 because they do not have a rigid cell wall and contain just one nucleus, one mitochondrion and one plastid, divisions of which can be highly synchronized by light/dark cycles5. This alga also has the smallest genome of all photosynthetic eukaryotes, and contains a minimal set of small membrane-bounded compartments; for example, a microbody (peroxisome), a single Golgi apparatus with two cisternae, coated vesicles, a single endoplasmic reticulum, and a few lysosome-like structures, as well as a small volume of cytosol (Fig. 1)6. One of the main points of interest that we discuss regarding this alga focuses on the origin, evolution and fundamental traits (for example, multiplication and differentiation) of single- as well as double-membrane-bounded organelles in plant cells. C. merolae, with its complete genomic information, provides an excellent opportunity for addressing such basic questions using microarray and proteome analyses. In addition, from an evolutionary perspective, C. merolae has other noteworthy properties that allow us to study the origin of eukaryotic cells, primary endosymbiosis between cyanobacteria and eukaryotic hosts, and secondary endosymbiosis between red algae and their hosts.
Figure 1: The unicellular red alga C. merolae 10D.
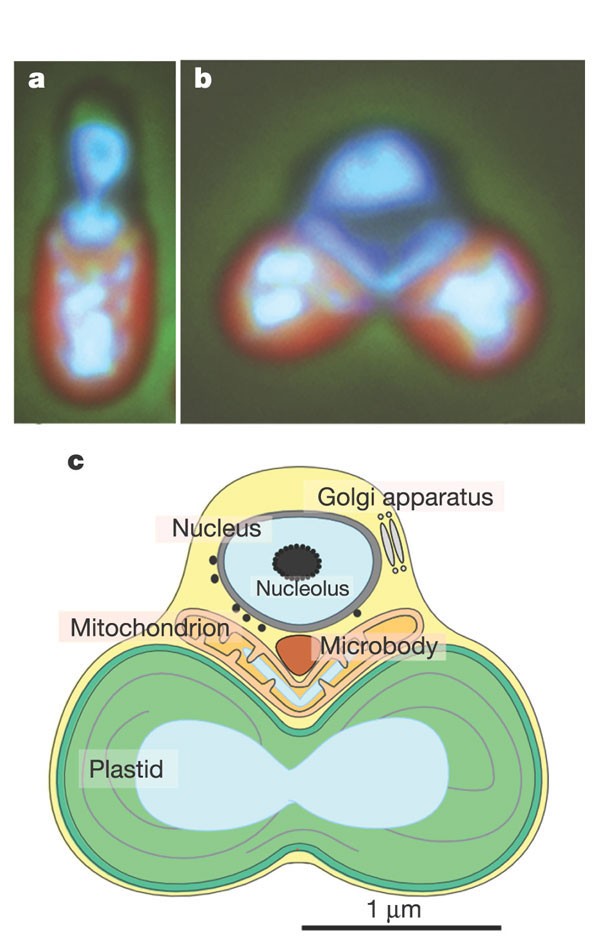
Phase contrast-fluorescent images of the interphase (a) and dividing (b) cells show localization of nuclear (top), mitochondrial (middle) and plastid DNA (bottom in blue/white) after DAPI staining. The plastids emit red autofluorescence. The schematic dividing cell (c) contains a nucleus, a V-shaped mitochondrion, a dumb-bell-shaped plastid, a microbody and a Golgi apparatus, divisions of which can be highly synchronized by light/dark cycles.
Samples of C. merolae 10D were isolated7 from the hot spring algal collection provided by G. Pinto (Naples University). The entire C. merolae genome was sequenced using the random sequencing method (see Methods). We obtained 16,520,305 base pairs (bp; approximately 99.98% of the estimated total length) of the nuclear genome sequence (Fig. 2, Table 1, and Supplementary Fig. 1 and Supplementary Table 1) with 46 gaps. The genome is distributed among the 20 chromosomes and ranges in size from approximately 0.42 to 1.62 Mb. No significant deviation in statistical parameters, such as base composition and gene density, were observed among the chromosomes (Supplementary Table 1). The overall G + C composition was 55.0%. The dinucleotide CpG in the C. merolae genome was exceptionally over-represented (1.151) compared with the expected value from observations of G + C content; it is generally underrepresented in other eukaryote genomes (Table 1).
Figure 2: Representation of C. merolae chromosomes.

a, Chromosome 1 and mapping of the BAC clones (50 kb and 100 kb). The chromosome is represented as a bar with pseudo-colour assignments of local G + C contents. Side rods represent genes and gene-like elements (left for transcription towards the top, and right for the bottom). Telomere and subtelomeric elements are indicated respectively as semicircles and purple rectangles at each end of the chromosome. b, A bird's-eye view of 20 chromosomes showing G + C contents, genes, subtelomeric elements (designations on the side: P, H, L, A, E, and so on), and RNA genes. A putative centromeric (A + T-rich) region is located on each chromosome.
Table 1 Nuclear and organellar genomes of C. merolae and comparison to other genomes
The putative repeat unit of telomeres in C. merolae is GGGGGGAAT, and as far as could be determined experimentally, it is found on both ends of the chromosomes. In addition, several sequence elements up to 20 kilobases (kb) in length were duplicated in 30 of the 40 putative subtelomeric regions (Fig. 2, Supplementary Fig. 1). Each chromosome has, in varying degrees, a single A + T-rich region on its mid-section. As chromosomal centromeric regions generally have a biased base composition, this A + T-rich region possibly defines centromeres (Fig. 2). The centromeres were confirmed via immunological experiments using antibodies against CENP-A, which was identified in the C. merolae genome (data not shown). Unlike many other eukaryotes, the C. merolae genome does not contain tandem repeated arrays of ribosomal RNA (rRNA) genes (Fig. 2). A single rRNA gene unit (18S-5.8S-28S) was discovered on three separate loci. The three units were virtually identical in sequence. Moreover, C. merolae has only three copies of the 5S rRNA gene, the sequences of which are also almost identical. Therefore, C. merolae has the smallest set of rRNA genes among all eukaryotes thus far studied. These results might be related to the existence of a single small nucleolus without nucleolus-associated chromatin. Furthermore, they also promote studies on the origin and formation of the nucleolus, because even prokaryotic cells with more than three copies of rRNA gene units do not have a nucleolus.
A full-length complementary DNA (cDNA) library was used to map expressed genes within the C. merolae genome. Fortunately, 99.85% of the expressed sequence tags (ESTs) were mapped on the genome sequence. In addition, many cDNA clones encoded a single open reading frame (ORF) bridging both end sequences. This suggests that most C. merolae genes lack introns. The predicted genes were automatically annotated using several databases (see Methods and Supplementary Information). As a result, 5,331 genes were identified, and 86.3% of them had corresponding ESTs (Supplementary Table 2). The number of genes in the C. merolae genome is similar to those found in yeasts and malarial parasites, despite the great ecological differences between these species (Table 1). Furthermore, the genes of the C. merolae genome are remarkable for their paucity of introns. Only 26 genes (0.5% of the protein genes) contained introns, and all but one of them had only a single intron. These introns had strict consensus sequences (Supplementary Fig. 2 and Supplementary Table 3).
Figure 3 summarizes the repertoire of C. merolae proteins on the basis of their assignment to eukaryotic clusters of orthologous groups (KOGs)8. Of the 4,771 predicted proteins, 2,536 were assigned to KOGs, by emulating the NCBI KOGnitor service (http://www.ncbi.nlm.nih.gov/COG/new/kognitor.html). The distribution of the functional classification of C. merolae was compared with those of other free-living unicellular eukaryotes, such as Saccharomyces cerevisiae9 and Schizosaccharomyces pombe10, and a higher plant Arabidopsis thaliana11. The distribution was on the whole similar to both yeasts which have similar genome size although C. merolae cells contain plastids. The lowered proportion of genes for ‘secondary metabolites biosynthesis, transport and catabolism’ found in these unicellular organisms, as compared with that of A. thaliana, might reflect their simple cellular organizations (Fig. 3, Supplementary Table 4).
Figure 3: Comparison of the functional classification of C. merolae proteins with other organisms.
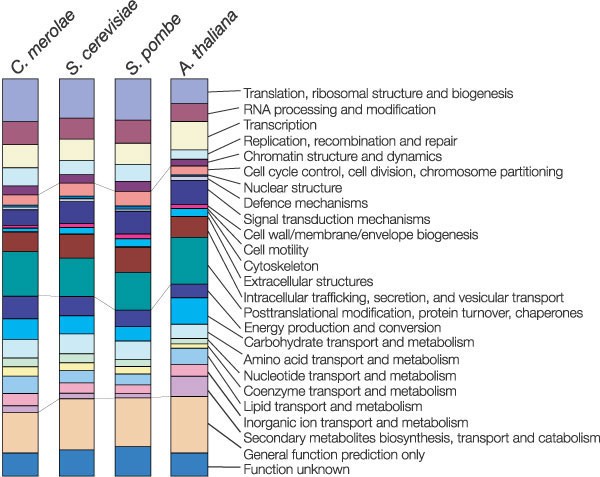
Columns represent the proportion of proteins assigned to KOG classification of each organism: C. merolae, S. cerevisiae, S. pombe and A. thaliana in a left-to-right fashion. The actual numbers of proteins assigned to each classification are given in Supplementary Table 3.
In C. merolae, the division of double-membrane-bounded mitochondria and plastids involves a dynamic trio: an FtsZ ring of bacterial origin, electron dense mitochondrial/plastid dividing rings (MD and PD rings), and eukaryotic mechanochemical dynamin rings. Four genes representing mitochondrial FtsZ (FtsZ2-1 and FtsZ2-2) and plastid FtsZ (FtsZ1-1 and FtsZ1-2) were identified12,13. A large gene family consisting of more than 10 members encoding functionally diverse dynamins with a wide range of membrane pinching roles have been found in other organisms; however, only two dynamin genes (C. merolae Dnm1 and Dnm2) are found, with a role in the later stages of the mitochondrion14 and plastid13 division, respectively. These findings suggest that plastids and mitochondria divide in a similar way, using very common systems consisting of the amalgamation of bacterial and eukaryotic rings. The dynamic trio of plastid division is conserved in lower algae to higher plants15,16. With mitochondrial divisions, however, whilst dynamin rings are retained in higher organisms, FtsZ and MD rings are not clearly observed, and it is possible that they were replaced by other systems during eukaryotic evolution13. MD/PD ring genes are yet unknown, although their identification should be accelerated by works such as this. Although the microbody, a single-membrane-bounded organelle, divides by binary fission in C. merolae17, it lacks Pex11p, which is a known key regulator of microbody division and proliferation18.
The following proteins related to cell motility and cytokinesis were encoded in C. merolae (Supplementary Table 5); one set of tubulin, two actins, five proteins of the kinesin family, and several intermediate filament proteins. However, no genes encoding myosin or proteins containing dynein motor domains were found. The absence of the myosin gene is consistent with the fact that electron microscopy and immuno-detection19 techniques did not detect microfilaments of actin; cDNA clones for actin genes were also not obtained. In the red alga Cyanidium caldarium RK-1,which is closely related to C. merolae but has a genome double the size7, cells divide using a contractile ring of actin filaments19. C. merolae cells therefore seem to divide using a system that is simpler than that of actomyosin.
C. merolae has noteworthy properties, which are relevant for examining the origin of eukaryotes, and primary and secondary plastid endosymbiosis20. Only 30 transfer RNAs (tRNAs) were detected in the nuclear genome using the program tRNAscan-SE with relaxed parameter settings (Fig. 2). Some of these tRNA genes showed possible archaeal features, namely, ectopic introns and anticodon GAU for tRNA-Ile (Supplementary Fig. 3). Four of these tRNA genes seemed to have introns in the D-loop region, whereas the introns of eukaryotic tRNA genes are limited to a site 3′ to the anticodon. As ectopic tRNA introns have been reported in some archaeal genomes21, this could explain the paucity of detected tRNA genes in C. merolae; tRNAscan-SE might have overlooked other tRNAs owing to the existence of unknown types of ectopic introns. Another point to note is that C. merolae possesses a single tRNA-Ile with anticodon GAU, which has not been observed in eukaryotes, but only in prokaryotes21.
Standard sets of photosystem genes, including those encoding phycobilisome components, were observed in C. merolae. Many of them (11 PSI genes and 17 PSII genes) are encoded in the plastid genome22, while PsbO, P, U, Z as well as a distant PsbQ homologue are encoded in the nuclear genome. Although only PsbU and PsbZ were previously identified in red algal PSII, the localization of PsbP and putative PsbQ in PSII, as recently suggested in Synechocystis sp. PCC680323 is an interesting subject of proteomic study. The genes psaH, N, X, as well as psbS and ndh genes are not encoded in either the plastid or the nuclear genomes. Therefore, the photosystems of C. merolae lack various mechanisms for dissipating excessive light energy.
Enzymes of the Calvin cycle in plants are known to be a mosaic of enzymes originating from cyanobacteria-like ancestors of an endosymbiont and its eukaryotic host24. Red algal ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) is known to be a product of horizontal gene transfer25. The origin of other Calvin cycle enzymes is essentially identical in C. merolae and A. thaliana (Supplementary Fig. 4 and Supplementary Table 6). It is highly probable that the complex and mosaic origin of Calvin cycle enzymes derived from common ancestors of green plants and red algae, and no essential changes occurred after the separation of the two lineages. This is strong support for the concept of a single event of primary plastid endosymbiosis. Among the known translocon proteins of plastids, Toc34, Toc75, Tic20, Tic22 and Tic110 were encoded in the C. merolae genome, but other proteins such as Toc159, Tic40 and Tic55 were not found. Results of phylogenetic analysis of the five translocon components (to be published elsewhere) also suggest the concept26.
Another aspect of the comparative genomics of the red algal genome is secondary endosymbiosis. Cryptophytes are thought to retain a remnant of the endosymbiotic red algal nucleus, the nucleomorph, in the periplastidic compartment. The sequencing of the cryptophyte alga Guillardia theta nucleomorph genome revealed a number of curious architectural features that might be shared by the genome of red algae27. C. merolae chromosomes showed multiple subtelomeric duplications, but did not contain rRNA gene clusters such as those of the nucleomorph genome. This implies that the telomeric rRNA gene clusters observed in the nucleomorph genome, as well as other prominent genome structures such as overlapped genes, appeared after secondary symbiosis. It is also notable that ectopic tRNA introns are also reported in nucleomorph tRNAs28. Details of the comparisons with the nucleomorph genome will be presented elsewhere.
Light signal transduction is critical for the growth and differentiation of photoautotrophic organisms. As the division of C. merolae cells is synchronized by light, an elaborate mechanism for light signal transduction must exist. Several putative blue light receptor (cryptochrome) genes were found in C. merolae, whereas no genes encoding phytochromes and phototropins were identified. As bacterial phytochrome genes are only found in some species of cyanobacteria with large genomes29, the ancestor of plastids might be an ancestral cyanobacterium without phytochromes. This also suggests that the phytochromes of higher plants might not be of cyanobacterial origin. In higher plants, various signalling pathways (such as the two-component system consisting of histidine kinases and response regulators as well as a MAP kinase cascade) are involved in the signal transduction of various hormones, and in the development of organs. In C. merolae, the presence of only a single candidate for histidine kinase and a dozen MAP kinase-related molecules is suggested. However, there are no response regulators other than those that are plastid-encoded, trimeric G protein and adenylate cyclase. Thus, C. merolae appear to use only a limited repertoire of signal transduction mechanisms, which corroborates the lack of cell differentiation in this alga.
C. merolae is an alga in which all of the three genome compartments—nucleus, mitochondrion (32,211 bp)30 and plastid (149,987 bp)22—have been sequenced. Such information is a prerequisite for future studies on proteomics, expression analysis using microarrays, and structural biology with heat-stable proteins that are unique among eukaryotes. All of this information will, in turn, help elucidate the origin, evolution and fundamental mechanisms of the single- as well as double-membrane-bounded organelles, and ultimately all photosynthetic eukaryotes. In addition, this hot spring alga will be useful in analysing the mechanisms of heat and acid tolerance in eukaryotic cells.