A high-resolution map of human evolutionary constraint using 29 mammals (original) (raw)
Abstract
The comparison of related genomes has emerged as a powerful lens for genome interpretation. Here we report the sequencing and comparative analysis of 29 eutherian genomes. We confirm that at least 5.5% of the human genome has undergone purifying selection, and locate constrained elements covering ∼4.2% of the genome. We use evolutionary signatures and comparisons with experimental data sets to suggest candidate functions for ∼60% of constrained bases. These elements reveal a small number of new coding exons, candidate stop codon readthrough events and over 10,000 regions of overlapping synonymous constraint within protein-coding exons. We find 220 candidate RNA structural families, and nearly a million elements overlapping potential promoter, enhancer and insulator regions. We report specific amino acid residues that have undergone positive selection, 280,000 non-coding elements exapted from mobile elements and more than 1,000 primate- and human-accelerated elements. Overlap with disease-associated variants indicates that our findings will be relevant for studies of human biology, health and disease.
Similar content being viewed by others
Main
A key goal in understanding the human genome is to discover and interpret all functional elements encoded within its sequence. Although only ∼1.5% of the human genome encodes protein sequence1, comparative analysis with the mouse2, rat3 and dog4 genomes showed that at least 5% is under purifying selection and thus probably functional, of which ∼3.5% consists of non-coding elements with probable regulatory roles. Detecting and interpreting these elements is particularly relevant to medicine, as loci identified in genome-wide association studies (GWAS) frequently lie in non-coding sequence5.
Although initial comparative mammalian studies could estimate the overall proportion of the genome under evolutionary constraint, they had little power to detect most of the constrained elements—especially the smaller ones. Thus, they focused only on the top 5% of constrained sequence, corresponding to less than ∼0.2% of the genome4,6. In 2005, we began an effort to generate sequence from a large collection of mammalian genomes with the specific goal of identifying and interpreting functional elements in the human genome on the basis of their evolutionary signatures7,8. Here we report our results to systematically characterize mammalian constraint using 29 eutherian (placental) genomes. We identify 4.2% of the human genome as constrained and ascribe potential function to ∼60% of these bases using diverse lines of evidence for protein-coding, RNA, regulatory and chromatin roles, and we present evidence of exaptation and accelerated evolution. All data sets described here are publicly available in a comprehensive data set at the Broad Institute and University of California, Santa Cruz (UCSC).
Sequencing, assembly and alignment
We generated genome sequence assemblies for 29 mammalian species selected to achieve maximum divergence across the four major mammalian clades (Fig. 1a and Supplementary Text 1 and Supplementary Table 1). For nine species, we used genome assemblies based on ∼7-fold coverage shotgun sequence, and for 20 species we generated ∼2-fold coverage (2×), to maximize the number of species sequenced with available resources on capillary machines. Twenty genomes are first reported here, and nine were previously described (see Supplementary Information).
Figure 1: Phylogeny and constrained elements from the 29 eutherian mammalian genome sequences.
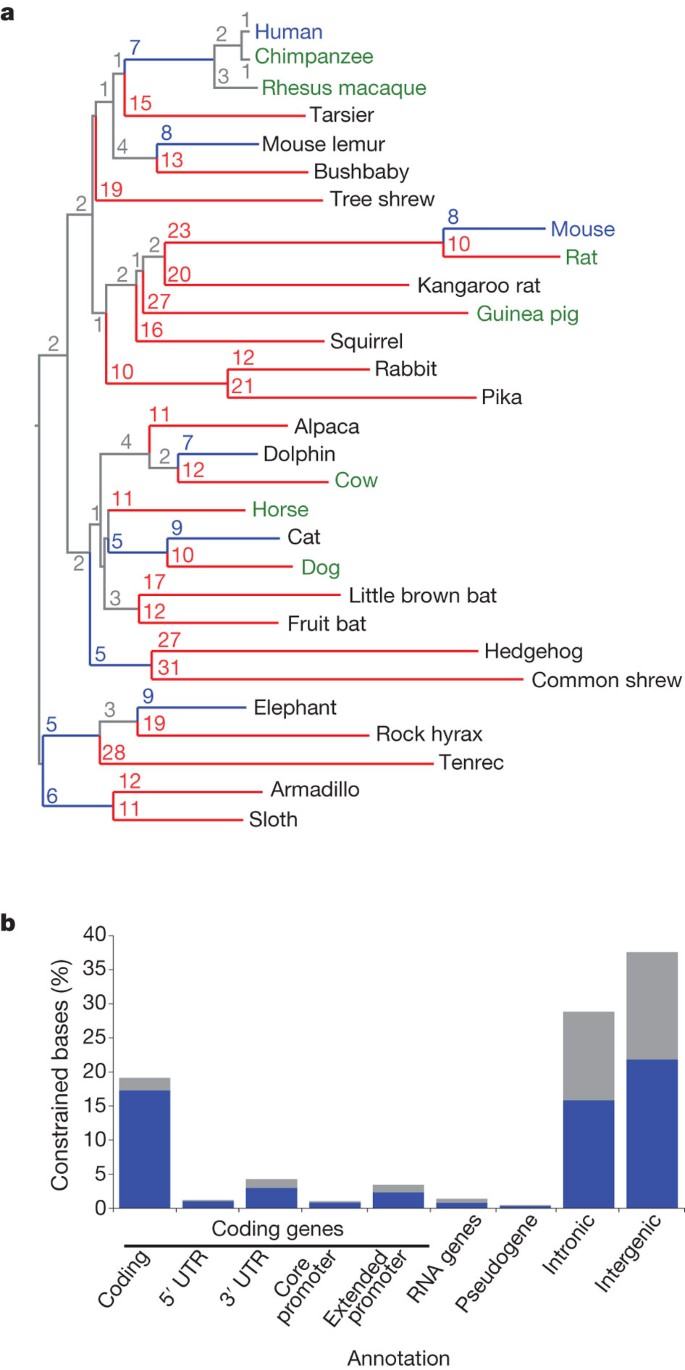
a, A phylogenetic tree of all 29 mammals used in this analysis based on the substitution rates in the MultiZ alignments. Organisms with finished genome sequences are indicated in blue, high quality drafts in green and 2× assemblies in black. Substitutions per 100 bp are given for each branch; branches with ≥10 substitutions are coloured red, blue indicates <10 substitutions. b, At 10% FDR, 3.6 million constrained elements can be detected encompassing 4.2% of the genome, including a substantial fraction of newly detected bases (blue) compared to the union of the HMRD 50-bp + Siepel vertebrate elements17 (see Supplementary Fig. 4b for comparison to HMRD elements only). The largest fraction of constraint can be seen in coding exons, introns and intergenic regions. For unique counts, the analysis was performed hierarchically: coding exons, 5′ UTRs, 3′ UTRs, promoters, pseudogenes, non-coding RNAs, introns, intergenic. The constrained bases are particularly enriched in coding transcripts and their promoters (Supplementary Fig. 4c).
The power to detect constrained elements depends largely on the total branch length of the phylogenetic tree connecting the species9. The 29 mammals correspond to a total effective branch length of ∼4.5 substitutions per site, compared to ∼0.68 for the human–mouse–rat–dog comparison (HMRD), and thus should offer greater power to detect evolutionary constraint: the probability that a genomic sequence not under purifying selection will remain fixed across all 29 species is _P_1 < 0.02 for single bases and _P_12 < 10−25 for 12-nucleotide sequences, compared to _P_1 ∼ 0.50 and _P_12 ∼ 10−3 for HMRD.
For mammals for which we generated 2× coverage, our assisted assembly approach10 resulted in a typical contig size N50C of 2.8 kb and a typical scaffold size N50S of 51.8 kb (Supplementary Text 2 and Supplementary Table 1) and high sequence accuracy (96% of bases had quality score Q20, corresponding to a <1% error rate)11. Compared to high-quality sequence across the 30 Mb of the ENCODE pilot project12, we estimated average error rates of 1–3 miscalled bases per kilobase11, which is ∼50-fold lower than the typical nucleotide sequence difference between the species, enabling high-confidence detection of evolutionary constraint (Supplementary Text 3).
We based our analysis on whole-genome alignments by MultiZ (Supplementary Text 4). The average number of aligned species was 20.9 at protein-coding positions in the human genome and 23.9 at the top 5% HMRD-conserved non-coding positions, with an average branch length of 4.3 substitutions per base in these regions (Supplementary Figs 1 and 2). In contrast, whole-genome average alignment depth is only 17.1 species with 2.9 substitutions per site, probably due to large deletions in non-functional regions4. The depth at ancestral repeats is 11.4 (Supplementary Fig. 1a), consistent with repeats being largely non-functional2,4.
Detection of constrained sequence
Our analysis did not substantially change the estimate of the proportion of genome under selection. By comparing genome-wide conservation to that of ancestral repeats, we estimated the overall fraction of the genome under evolutionary constraint to be 5.36% at 50-bp windows (5.44% at 12-bp windows), using the SiPhy-ω statistic13, a measure of overall substitution rate (Supplementary Fig. 3), consistent with previous similar estimates2,4,14. However, alternative methods15,16 and different ways of correcting for the varying alignment depths give higher estimates (see Supplementary Text 5 for details).
The additional species had a marked effect on our ability to identify the specific elements under constraint. With 29 mammals, we pinpoint 3.6 million elements spanning 4.2% of the genome, at a finer resolution of 12 bp (Fig. 1b and Supplementary Text 6, Supplementary Fig. 4, Supplementary Tables 2 and 3), compared to <0.1% of the genome for HMRD 12-bp elements and 2.0% for HMRD 50-bp elements4. Elements previously detected using five vertebrates17 also detect a larger fraction of the genome (∼4.1%), but only cover 45% of the mammalian elements detected here, suggesting that a large fraction of our elements are mammalian specific. The mean element size (36 bp) is considerably shorter than both previously detected HMRD elements (123 bp) and five-vertebrate elements (104 bp)17. For example, it is now possible to detect individual binding sites for the neuron-restrictive silencer factor (NRSF) in the promoter of the NPAS4 gene, which are beyond detection power in previous data sets (Fig. 2 and Supplementary Fig. 5). We found a similar regional distribution of 12-bp elements (including the 2.6 million newly detected constrained elements) to previously detected HMRD elements (r = 0.94, Supplementary Fig. 6). Similar results were obtained with the PhastCons17 statistic (see Supplementary Text 6).
Figure 2: Identification of four NRSF-binding sites in NPAS4.
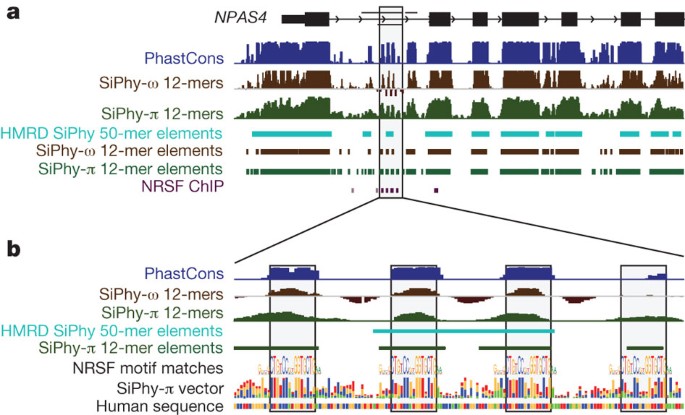
a, The neurological gene NPAS4 has many constrained elements overlapping introns and the upstream intergenic region. The grey shaded box contained only one constrained element using HMRD, whereas analysis of 29 mammalian sequences reveals four smaller elements. b, These four constrained elements in the first intron correspond to binding sites for the NRSF transcription factor, known to regulate neuronal lineages.
Using a new method, SiPhy-π, sensitive not just to the substitution rate but also to biases in the substitution pattern (for example, positions free to mutate between G and T only, Supplementary Fig. 7), we detected an additional 1.3% of the human genome in constrained elements (see Supplementary Tables 2 and 3). Most of the newly detected constrained nucleotides extend elements found by rate-based methods, but 22% of nucleotides lie in new elements (average length 17 bp) and are enriched in non-coding regions.
Constraint within the human population
We observed that the evolutionary constraint acting on the 29 mammals is correlated with constraint within the human population, as assessed from human polymorphism data (Supplementary Text 7) and consistent with previous studies18. Mammalian constrained elements show a depletion in single-nucleotide polymorphisms (SNPs)19, and more constrained elements show even greater depletion. For example, in the top 1% most strongly conserved non-coding regions, SNPs occur at a 1.9-fold lower rate than the genome average, and the derived alleles have a lower frequency, consistent with purifying selection at many of these sites in the human population.
Moreover, at positions with biased substitution patterns across mammals, the observed human SNPs show a similar bias to the one observed across mammals (Supplementary Fig. 7). Thus, not only are constrained regions less likely to exhibit polymorphism in humans, but when such polymorphisms are observed, the derived alleles in humans tend to match the alleles present in non-human mammals, indicating a preference for the same alleles across both mammalian and human evolution.
Functional annotation of constraint
We first studied the overlap of the 3.6 million evolutionarily constrained elements (ω < 0.8 and P < 10−15) with known gene annotations (Fig. 1b). Roughly 30% of constrained elements were associated with protein-coding transcripts: ∼25.3% overlap mature messenger RNAs (including 19.6% in coding exons, 1.2% in 5′ untranslated regions (5′ UTRs) and 4.4% in 3′ UTRs), and an additional 4.4% reside within 2 kb of transcriptional start sites (1.2% reside within 200 bases).
The majority of constrained elements, however, reside in intronic and intergenic regions (29.7% and 38.6%, respectively). To study their biological roles and provide potential starting points to understand these large and mostly uncharted territories, we next studied their overlap with evolutionary signatures7,8,20,21 characteristic of specific types of features and a growing collection of public large-scale experimental data.
Protein-coding genes and exons
Despite intense efforts to annotate protein-coding genes over the past decade20,22,23,24, we detected 3,788 candidate new exons (a 2% increase) using evolutionary signatures characteristic of protein-coding exons25. Of these, 54% reside outside transcripts of protein-coding genes, 19% within introns, and 13% in UTRs of known coding genes (Supplementary Text 8, Supplementary Tables 4 and 5). Our methods recovered 92% of known coding exons that were larger than 10 codons and fall in syntenic regions, the remainder showing non-consensus splice sites, unusual features, or poor conservation.
The majority of new exon candidates (>58%) are supported by evidence of transcription measured in 16 human tissues26 (Supplementary Fig. 8a) or similarity to known Pfam protein domains. Thirty-one per cent of intronic and 13% of intergenic predictions extend known transcripts, and 5% and 11% respectively reside in new transcript models. The newly detected exons are more tissue specific than known exons (mean of 3 tissues versus 12) and are expressed at fivefold lower levels. Directed experiments and manual curation will be required to complete the annotation of the few hundred protein-coding genes that probably remain unannotated27.
We found apparent stop codon readthrough28 of four genes based on continued protein-coding constraint after an initial conserved stop codon29 and until a subsequent stop codon (Supplementary Text 9 and Supplementary Fig. 8b). Readthrough in SACM1L could be triggered by an 80-base conserved RNA stem loop predicted by RNAz30, lying four bases downstream of the readthrough stop codon.
We also detected coding regions with a very low synonymous substitution rate, indicating additional sequence constraints beyond the amino acid level (Supplementary Text 9). We found >10,000 such synonymous constraint elements (SCEs) in more than one-quarter of all human genes31. Initial analysis indicates potential roles in splicing regulation (34% span an exon–exon junction), A-to-I editing, microRNA (miRNA) targeting and developmental regulation. HOX genes contain several top candidates (Fig. 3a), including two previously validated developmental enhancers32,33.
Figure 3: Examination of evolutionary signatures identifies SCEs and evidence of positive selection.

a, Two regions within the HOXA2 open reading frame are identified as SCEs (red), corresponding to overlapping functional elements within coding regions. Note that the synonymous rate reductions are not obvious from the base-wise conservation measure (in blue). Both elements have been characterized as enhancers driving HOXA2 expression in distinct segments of the developing mouse hindbrain. The element in the first exon encodes Hox–Pbx-binding sites and drives expression in rhombomere 4 (ref. 33), whereas the element in the second exon contains Sox-binding sites and drives expression in rhombomere 2 (ref. 32). Synonymous constraint elements are also found in most other HOX genes, and up to a quarter of all genes. b, Although ∼85% of genes show only negative (purifying) selection and 9% of genes show uniform positive selection, the remaining 6% of genes, including ABI2, show only localized regions of positively selected sites. Each vertical bar covers the estimated 95% confidence interval for dN/dS at that site (with values of 0 truncated to 0.01 to accommodate the log scaling), and bars are coloured according to a signed version of the SLR statistic for non-neutral evolution: blue for sites under purifying selection, grey for neutral sites and red for sites under positive selection.
RNA structures and families of structural elements
We next used evolutionary signatures characteristic of conserved RNA secondary structures34 to reveal 37,381 candidate structural elements (Supplementary Text 10 and Supplementary Fig. 9a), covering ∼1% of constrained regions. For example, the XIST large intergenic non-coding RNA (lincRNA), known to bind chromatin and enable X-chromosome inactivation35, contains a newly predicted structure in its 3′ end (Supplementary Fig. 9b, f)—distinct from other known structures36—which seems to be the source of chromatin-associated short RNAs37.
Sequence- and structure-based clustering of predictions outside protein-coding exons revealed 1,192 novel families of structural RNAs (Supplementary Text 10). We focused on a high-scoring subset consisting of 220 families with 725 instances, which also showed the highest thermodynamic stability30 (Supplementary Figs 9a and 10), DNase hypersensitivity, expression pattern correlation across tissues and intergenic expression enrichment (Supplementary Fig. 9a). We also expanded both known and novel families by including additional members detected by homology to existing members.
Noteworthy examples include: a glycyl-tRNA family, including a new member in POP1, involved in tRNA maturation and probably involved in feedback regulation of POP1; three intronic families of long hairpins in ion-channel genes known to undergo A-to-I RNA editing and possibly involved in regulation of the editing event; an additional member of a family of 5′ UTR hairpins overlapping the start codon of collagen genes and potential new miRNA genes that extend existing families37.
Two of the largest novel families consist of short AU-rich hairpins of 6–7 bp that share the same strong consensus motif in their stem. These occur in the 3′ UTRs of genes in several inflammatory response pathways, the post-transcriptional regulation of which often involves AU-rich elements (AREs). Indeed, two homologous hairpins in TNF and CSF3 correspond to known mRNA-destabilization elements, suggesting roles in mRNA stability for the two families37.
Lastly, a family of six conserved hairpin structures (Supplementary Fig. 9g) was found in the 3′ UTR of the MAT2A gene37, which is involved in the synthesis of _S_-adenosylmethionine (SAM), the primary methyl donor in human cells. All six hairpins consist of a 12–18-bp stem and a 14-bp loop region with a deeply conserved sequence motif (Supplementary Fig. 9e), and may be involved in sensing SAM concentrations, which are known to affect MAT2A mRNA stability38.
Conservation patterns in promoters
As different types of conservation in promoters may imply distinct biological functions39, we classified the patterns of conservation within core promoters into three categories: (1) those with uniformly ‘high’ constraint (7,635 genes, 13,996 transcripts); (2) uniformly ‘low’ constraint (2,879 genes, 4,135 transcripts); and (3) ‘intermittent’ constraint, consisting of alternating peaks and troughs of conservation (14,271 genes and 29,814 transcripts) (Supplementary Fig. 11a). High and intermittent constraint promoters are both associated with CpG islands (∼66%), whereas low constraint promoters have significantly lower overlap (∼41%), and all three classes show similar overlap with functional TATA boxes (2–3%, see Supplementary Text 11).
These groups show distinct Gene Ontology enrichments (Supplementary Fig. 11b), with high-constraint promoters involved in development (P with Bonferroni correction (_P_Bonf) < 10−30), intermittent constraint in basic cellular functions (_P_Bonf < 5 × 10−4), and low-constraint promoters in immunity, reproduction and perception, functions expected to be under positive selection and lineage-specific adaptation2.
High constraint may reflect cooperative binding of many densely binding factors, as previously suggested for developmental genes6. Intermittent constraint promoters, the peak-spacing distribution of which was suggestive of the periodicity of the DNA helix turns, may reflect loosely interacting factors (Supplementary Fig. 11c, d). Low constraint may reflect rapid motif turnover, under neutral drift or positive selection.
Identifying specific instances of regulatory motifs
Data from just four species (HMRD) was sufficient to create a catalogue of known and novel motifs with many conserved instances across the genome21. The power to discover such motifs was high, because one can aggregate data across hundreds of motif instances. Not surprisingly, the additional genomes therefore had little effect on the ability to discover new motifs (known motifs showed 99% correlation in genome-wide motif conservation scores, Supplementary Figs 12 and 13).
In contrast, the 29 mammalian genomes markedly improved our ability to detect individual motif instances, making it possible to predict specific target sites for 688 regulatory motifs corresponding to 345 transcription factors (Supplementary Fig. 14). We chose to identify motif instances at a false discovery rate (FDR) of 60%, representing a reasonable compromise between specificity and sensitivity given the available discovery power (Supplementary Text 12), and matching the experimental specificity of chromatin immunoprecipitation (ChIP) experiments for identifying biologically significant targets40. Higher levels of stringency could be obtained by sequencing additional species.
We identified 2.7 million conserved instances (Supplementary Table 6), enabling the construction of a regulatory network linking 375 motifs to predicted targets, with a median of 21 predicted regulators per target gene (25th percentile, 10; 75th percentile, 39). The number of target sites (average, 4,277; 25th percentile, 1,407; 75th percentile, 10,782) are comparable to those found in ChIP experiments, and have the advantage that they are detected at nucleotide resolution, enabling us to use them to interpret disease-associated variants for potential regulatory functions. However, some motifs never reached high confidence values, and others did so at very few instances.
The motif-based targets show strong agreement with experimentally defined binding sites from ChIP experiments (Supplementary Table 7). For long and distinct motifs, such as CTCF and NRSF, the fraction of instances overlapping experimentally observed binding matches the fraction predicted by the confidence score (for example, at 80% confidence 70% of NRSF motif instances overlapped bound sites, and at ∼50% confidence 40% overlapped), despite potential confounding aspects such as condition-specific binding, overlapping motifs between factors, or non-specific binding. Moreover, increasing confidence levels showed increasing overlap with experimental binding (Supplementary Figs 14–16). For example, YY1 enrichment for bound sites increased from 42-fold to 168-fold by focusing on conserved instances. Lastly, combining motif conservation and experimental binding led to increased enrichment for candidate tissue-specific enhancers, suggesting that the two provide complementary information. Within bound regions, the evolutionary signal reveals specific motif instances with high precision (for example, Figs 2 and 4 and Supplementary Fig. 17).
Figure 4: Using constraint to identify candidate mutations.
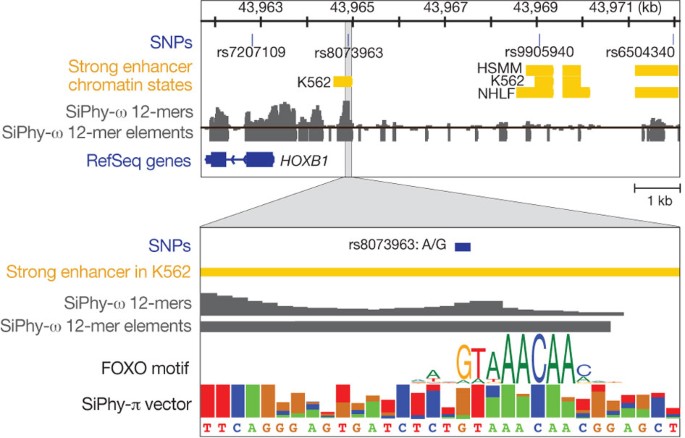
Conservation can help us resolve amid multiple SNPs the ones that disrupt conserved functional elements and are likely to have regulatory roles. In this example, a SNP (rs6504340) associated with tooth development is strongly linked to a conserved intergenic SNP, rs8073963, 7.1 kb away, which disrupts a deeply conserved Forkhead-family motif in a strong enhancer. Although the SNPs shown here stem from GWAS on HapMap data, the same principle should be applicable to associated variants detected by resequencing the region of interest.
Chromatin signatures
To suggest potential functions for the ∼68% of ‘unexplained’ constrained elements outside coding regions, UTRs or proximal promoters, we used chromatin state maps from CD4 T cells41 (Supplementary Fig. 18) and nine diverse cell types42 (Supplementary Text 13 and Supplementary Fig. 19). In T cells, constrained elements were most enriched for promoter-associated states (up to fivefold), an insulator state and a specific repressed state (2.2-fold), and numerous enhancer states (1.5–2-fold), together covering 7.1% of the unexplained elements at 2.1-fold enrichment. In the nine cell types, enriched promoter, enhancer and insulator states cover 36% of unexplained elements at ∼1.75-fold enrichment, with locations active in multiple cell types showing even stronger enrichment (Supplementary Fig. 20).
Overall, chromatin states indicate possible functions (at 1.74-fold enrichment) for 37.5% (N = 987,985) of unexplained conserved elements (27% of all conserved elements), suggesting meaningful association for at least 16% of unexplained constrained bases. Although current experiments only provide nucleosome-scale (∼200-bp) resolution, we expect higher-resolution experimental assays that more precisely pinpoint regulatory regions to show further increases in enrichment. The increased overlap observed with additional cell types suggests that new cell types will help elucidate additional elements. Of course, further experimental tests will be required to validate the predicted functional roles.
Accounting for constrained elements
Overall, ∼30% of constrained elements overlap were associated with protein-coding transcripts, ∼27% overlap specific enriched chromatin states, ∼1.5% novel RNA structures, and ∼3% conserved regulatory motif instances (Supplementary Text 13, 14). Together, ∼60% of constrained elements overlap one of these features, with enrichments ranging from 1.75-fold for chromatin states (compared to unannotated regions) up to 17-fold for protein-coding exons (compared to the whole genome).
Implications for interpreting disease-associated variants
In the non-protein-coding genome, SNPs associated with human diseases in genome-wide association studies are 1.37-fold enriched for constrained regions, relative to HapMap SNPs (Supplementary Text 15 and Supplementary Table 8). This is notable because only a small proportion of the associated SNPs are likely to be causative, whereas the rest are merely in linkage disequilibrium with causative variants.
Accordingly, constrained elements should be valuable in focusing the search for causative variants among multiple variants in linkage disequilibrium. For example, in an intergenic region between HOXB1 and HOXB2 associated with tooth development phenotypes43, the reported SNP (rs6504340) is not conserved, but a linked SNP (rs8073963) sits in a constrained element 7.1 kb away. Moreover, rs8073963 disrupts a deeply conserved FOXO2 motif instance within a predicted enhancer (Fig. 4), making it a candidate mutation for further follow-up. Similar examples of candidate causal variants are found for diverse phenotypes such as height or multiple sclerosis, and similar analyses could be applied to case–control resequencing data.
Evolution of constrained elements
We next sought to identify signatures of positive selection that may accompany functional adaptations of different species to diverse environments and new ecosystems.
Codon-specific selection
We used the ratio dN/dS of non-synonymous to synonymous codon substitutions as evidence of positive selection (>1) or negative selection (<1). Although dN/dS is typically calculated for whole genes, the additional mammals sequenced enabled analysis at the codon level: simulations predicted a 250-fold gain in sensitivity compared to HMRD, identifying 53% of positive sites at 5% FDR (Supplementary Text 16).
Applying this test to 6.05 million codons in 12,871 gene trees, we found evidence of strong purifying selection (dN/dS < 0.5) for 84.2% of codons and positive selection (dN/dS > 1.5) for 2.4% of codons (with 94.1% of sites <1 and 5.9% >1; Supplementary Table 9). At 5% FDR, we found 15,383 positively selected sites in 4,431 proteins. The genes fall into three classes based on the distribution of selective constraint: 84.8% of genes show uniformly high purifying selection, 8.9% show distributed positive selection across their length and 6.3% show localized positive selection concentrated in small clusters (Fig. 3b and Supplementary Fig. 21, Supplementary Tables 10 and 11).
Genes with distributed positive selection were enriched in such functional categories as immune response (_P_Bonf < 10−16) and taste perception (_P_Bonf < 10−10), which are known to evolve rapidly, but also in some unexpected functions such as meiotic chromosome segregation (_P_Bonf < 10−23) and DNA-dependent regulation of transcription (_P_Bonf < 10−19; Supplementary Table 12). Localized positive selection was enriched in core biochemical processes, including microtubule-based movement (_P_Bonf < 10−10), DNA topological change (_P_Bonf < 10−4) and telomere maintenance (_P_Bonf < 7 × 10−3), suggesting adaptation at important functional sites.
Focusing on 451 unique Pfam protein-domain annotations, we found abundant purifying selection, with 225 domains showing purifying selection for >75% of their sites, and 447 domains showing negative selection for >50% of their sites (Supplementary Table 13). Domains with substantial fractions of positively selected sites include CRAL/TRIO involved in retinal binding (2.6%), proteinase-inhibitor-cystatin involved in bone remodelling (2.2%) and the secretion-related EMP24/GOLD/P24 family (1.6%).
Exaptation of mobile elements
Mobile elements provide an elegant mechanism for distributing a common sequence across the genome, which can then be retained in locations where it confers advantageous regulatory functions to the host—a process termed exaptation. Our data revealed >280,000 mobile element exaptations common to mammalian genomes covering ∼7 Mb (Supplementary Text 17), a considerable expansion from the ∼10,000 previously recognized cases44. Of the ∼1.1 million constrained elements that arose during the 90 million years between the divergence from marsupials and the eutherian radiation, we can trace >19% to mobile element exaptations. Often only a small fraction (median ∼11%) of each mobile element is constrained, in some cases matching known regulatory motifs. Recent exaptations are generally found near ancestral regulatory elements, except in gene deserts, which are abundant in ancestral elements but show few recent exaptations (P < 10−300, Supplementary Fig. 22).
Accelerated evolution in the primate lineage
Lineage-specific rapid evolution in ancestrally constrained elements previously revealed human positive selection associated with brain and limb development45. Applying this signature to the human and primate lineages, we identified 563 human-accelerated regions (HARs) and 577 primate-accelerated regions (PARs) at FDR <10% (Supplementary Text 18, Supplementary Tables 14 and 15), significantly expanding the 202 previously known HARs46. Fifty-four HARs (9.4%) and 49 PARs (8.5%) overlap enhancer-associated chromatin marks and experimentally validated enhancers (Supplementary Text 18). Substitution patterns in HARs suggest that GC-biased gene conversion (BGC) is not responsible for the accelerated evolution in the vast majority of these regions (∼15% show evidence of BGC).
Genes harbouring or neighbouring HARs and PARs are enriched for extracellular signalling, receptor activity, immunity, axon guidance, cartilage development and embryonic pattern specification (Supplementary Fig. 23). For example, the FGF13 locus associated with an X-linked form of mental retardation contains four HARs near the 5′ ends of alternatively spliced isoforms of FGF13 expressed in the nervous system, epithelial tissues and tumours, suggesting human-specific changes in isoform regulation (Supplementary Fig. 24).
Discussion
Comparative analysis of 29 mammalian genomes reveals a high-resolution map of >3.5 million constrained elements that encompass ∼4% of the human genome and suggest potential functional classes for ∼60% of the constrained bases; the remaining 40% show no overlap and remain uncharacterized. We report previously undetected exons and overlapping functional elements within protein-coding sequence, new classes of RNA structures, promoter conservation profiles and predicted targets of transcriptional regulators. We also provide evidence of evolutionary innovation, including codon-specific positive selection, mobile element exaptation and accelerated evolution in the primate and human lineages.
By focusing our comparison on only eutherian mammals, we discover functional elements relevant to this clade, including recent eutherian innovations. This is especially important for discovering regulatory elements, which can be subject to rapid turnover47. Indeed, a previous comparison indicated that only 80% of 50-bp non-coding elements are shared with opossum48, and the current 12-bp analysis shows ∼64% of non-coding elements shared with opossum, and only 6% with stickleback fish. Many eutherian elements are thus probably missing from previous maps of vertebrate constraint17.
Sequencing of additional species should enable discovery of lineage-specific elements within mammalian clades, and provide increased resolution for shared mammalian constraint. We estimate that 100–200 eutherian mammals (15–25 neutral substitutions per site) will enable single-nucleotide resolution. The majority of this branch length is present within the Laurasiatherian and Euarchontoglire branches, which also contain multiple model organisms. These are ideal next targets for sequencing as part of the Genome 10K effort49, aiming to sequence 10,000 vertebrate species. Within the primate clade, a branch length of ∼1.5 could probably be achieved, enabling primate-specific selection studies, albeit at lower resolution. Lastly, human-specific selection should be detectable by combining data across genomic regions and by comparing thousands of humans50.
The constrained elements reported here can be used to prioritize disease-associated variants for subsequent study, providing a powerful lens for elucidating functional elements in the human genome complementary to ongoing large-scale experimental endeavours such as ENCODE and Roadmap Epigenomics. Experimental studies require prior knowledge of the biochemical activity sought and reveal regions active in specific cell types and conditions. Comparative approaches provide an unbiased catalogue of shared functional regions independent of biochemical activity or condition, and thus can capture experimentally intractable or rare activity patterns. With increasing branch length, they can provide information on ancestral and recent selective pressures across mammalian clades and within the human population. Ultimately, the combination of disease genetics, comparative and population genomics and biochemical studies have important implications for understanding human biology, health and disease.
Methods Summary
A full description of materials and methods, including sample selection and sequencing strategy, assembly strategies and results, error estimation and correction, alignment details, estimation of genome portion under constraint, detection of constrained elements, mammalian constraint versus human polymorphism, protein coding genes, detection of stop codon readthrough and synonymous constraint elements, RNA structure detection, patterns of promoter constraint, regulatory motif discovery, correlation with chromatin state information, overall accounting of constraint elements, comparison with disease-associated variants, detection of codon-specific positive selection, exaptation of ancestral repeat elements, and human and primate accelerated regions is available in Supplementary Information. All animal experiments were approved by the MIT Committee for Animal Care.
Accession codes
Data deposits
A complete set of data files can be downloaded from the Broad website (https://www.broadinstitute.org/scientific-community/science/projects/mammals-models/29-mammals-project-supplementary-info) or viewed using the UCSC Genome Browser (http://genomewiki.cse.ucsc.edu/index.php/29mammals) or the Broad Institute Integrative Genome Viewer (http://www.broadinstitute.org/igv/projects/29mammals). NCBI accession numbers for all newly sequenced genomes can be found in Supplementary Table 1.
References
- Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001)
Article ADS CAS Google Scholar - Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002)
Article ADS CAS Google Scholar - Gibbs, R. A. et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521 (2004)
Article ADS CAS Google Scholar - Lindblad-Toh, K. et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819 (2005)
Article ADS CAS Google Scholar - Altshuler, D., Daly, M. J. & Lander, E. S. Genetic mapping in human disease. Science 322, 881–888 (2008)
Article ADS CAS Google Scholar - Bejerano, G. et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004)
Article ADS CAS Google Scholar - Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423, 241–254 (2003)
Article ADS CAS Google Scholar - Stark, A. et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450, 219–232 (2007)
Article ADS CAS Google Scholar - Cooper, G. M., Brudno, M., Green, E. D., Batzoglou, S. & Sidow, A. Quantitative estimates of sequence divergence for comparative analyses of mammalian genomes. Genome Res. 13, 813–820 (2003)
Article CAS Google Scholar - Gnerre, S., Lander, E. S., Lindblad-Toh, K. & Jaffe, D. B. Assisted assembly: how to improve a de novo genome assembly by using related species. Genome Biol. 10, R88 (2009)
Article Google Scholar - Hubisz, M. J., Lin, M. F., Kellis, M. & Siepel, A. Error and error mitigation in low-coverage genome assemblies. PLoS ONE 14, e17034 (2011)
Article Google Scholar - Thomas, J. W. et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424, 788–793 (2003)
Article ADS CAS Google Scholar - Garber, M. et al. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25, i54–i62 (2009)
Article ADS CAS Google Scholar - Chiaromonte, F. et al. The share of human genomic DNA under selection estimated from human-mouse genomic alignments. Cold Spring Harb. Symp. Quant. Biol. 68, 245–254 (2003)
Article CAS Google Scholar - Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025 (2010)
Article Google Scholar - Meader, S., Ponting, C. P. & Lunter, G. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 20, 1335–1343 (2010)
Article CAS Google Scholar - Siepel, A. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005)
Article CAS Google Scholar - Drake, J. A. et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nature Genet. 38, 223–227 (2006)
Article CAS Google Scholar - Keinan, A., Mullikin, J. C., Patterson, N. & Reich, D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nature Genet. 39, 1251–1255 (2007)
Article CAS Google Scholar - Clamp, M. et al. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl Acad. Sci. USA 104, 19428–19433 (2007)
Article ADS CAS Google Scholar - Xie, X. et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434, 338–345 (2005)
Article ADS CAS Google Scholar - Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008)
Article ADS CAS Google Scholar - Siepel, A. et al. Targeted discovery of novel human exons by comparative genomics. Genome Res. 17, 1763–1773 (2007)
Article CAS Google Scholar - Pruitt, K. D. et al. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 19, 1316–1323 (2009)
Article CAS Google Scholar - Lin, M. F., Jungreis, I. & Kellis, M. PhyloCSF: a comparative genomics method to distinguish protein-coding and non-coding regions. Bioinformatics 27, i275–i282 (2010)
Article Google Scholar - Guttman, M. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature Biotechnol. 28, 503–510 (2010)
Article CAS Google Scholar - Harrow, J. et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 7, (suppl. 1)1–9 (2006)
Article Google Scholar - Lin, M. F. et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Res. 17, 1823–1836 (2007)
Article CAS Google Scholar - Jungreis, I. et al. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 10.1101/gr.119974.110 (in the press)
- Washietl, S., Hofacker, I. L. & Stadler, P. F. Fast and reliable prediction of noncoding RNAs. Proc. Natl Acad. Sci. USA 102, 2454–2459 (2005)
Article ADS CAS Google Scholar - Lin, M. F. et al. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res. 10.1101/gr.108753.110 (in the press)
- Tumpel, S., Cambronero, F., Sims, C., Krumlauf, R. & Wiedemann, L. M. A regulatory module embedded in the coding region of Hoxa2 controls expression in rhombomere 2. Proc. Natl Acad. Sci. USA 105, 20077–20082 (2008)
Article ADS CAS Google Scholar - Lampe, X. et al. An ultraconserved Hox–Pbx responsive element resides in the coding sequence of Hoxa2 and is active in rhombomere 4. Nucleic Acids Res. 36, 3214–3225 (2008)
Article CAS Google Scholar - Pedersen, J. S. et al. Identification and classification of conserved RNA secondary structures in the human genome. PLOS Comput. Biol. 2, e33 (2006)
Article ADS CAS Google Scholar - Lee, J. T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 23, 1831–1842 (2009)
Article CAS Google Scholar - Maenner, S. et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol. 8, e1000276 (2010)
Article Google Scholar - Parker, B. J. et al. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 10.1101/gr.112516.110 (in the press)
- Martinez-Chantar, M. L. et al. l-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: role of _S_-adenosylmethionine. J. Biol. Chem. 278, 19885–19890 (2003)
Article Google Scholar - Baek, D., Davis, C., Ewing, B., Gordon, D. & Green, P. Characterization and predictive discovery of evolutionarily conserved mammalian alternative promoters. Genome Res. 17, 145–155 (2007)
Article CAS Google Scholar - Kheradpour, P., Stark, A., Roy, S. & Kellis, M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 17, 1919–1931 (2007)
Article CAS Google Scholar - Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature Biotechnol. 28, 817–825 (2010)
CAS Google Scholar - Ernst, J. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011)
Article ADS CAS Google Scholar - Pillas, D. et al. Genome-wide association study reveals multiple loci associated with primary tooth development during infancy. PLoS Genet. 6, e1000856 (2010)
Article Google Scholar - Lowe, C. B., Bejerano, G. & Haussler, D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc. Natl Acad. Sci. USA 104, 8005–8010 (2007)
Article ADS CAS Google Scholar - Prabhakar, S. et al. Human-specific gain of function in a developmental enhancer. Science 321, 1346–1350 (2008)
Article ADS CAS Google Scholar - Pollard, K. S. et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2, e168 (2006)
Article Google Scholar - Schmidt, D. et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040 (2010)
Article ADS CAS Google Scholar - Mikkelsen, T. S. et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447, 167–177 (2007)
Article ADS CAS Google Scholar - Genome 10K Community Of Scientists Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered. 100, 659–674 (2009)
Article Google Scholar - A. map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010)
Acknowledgements
We thank O. Ryder, E. Fuchs, D. Haring, A. Walsh, D. Duffield, S. Wong, T. Alvarado, J. Boylan, S. Combes, P. deJong, J. Allman, J. Patton, D. McMullen, D. Hafner, D. Miller, T. Kunz, G. Hewitt, J. Searle, H. Künzle and D. Williams for providing organismal material. We thank L. Gaffney for help with figures. This work was supported by the National Human Genome Research Institute (NHGRI), including grant U54 HG003273 (R.A.G.), National Institute for General Medicine (NIGMS) grant no. GM82901 (Pollard laboratory) and the European Science Foundation (EURYI award to K.L.-T.), NSF National Science Foundation (NSF) postdoctoral fellowship award 0905968 (J.E.), National Science Foundation CAREER 0644282 and NIH R01 HG004037 and the Sloan Foundation (M.K.), and an Erwin Schrödinger Fellowship of the Austrian Fonds zur Förderung der Wissenschaftlichen Forschung (S.W.), the Gates Cambridge Trust (G.J.), Novo Nordisk Foundation (B.J.P. and J.W.); a Statistics Network Fellowship, Department of Mathematical Sciences, University of Copenhagen (B.J.P.); the David and Lucile Packard Foundation (A.S.); the Danish Council for Independent Research Medical Sciences (J.S.P.); The Lundbeck Foundation (J.S.P.).
Author information
Author notes
- Manuel Garber, Or Zuk, Michael F. Lin, Brian J. Parker, Stefan Washietl, Pouya Kheradpour, Jason Ernst, Gregory Jordan, Evan Mauceli, Lucas D. Ward, Craig B. Lowe, Alisha K. Holloway, Michele Clamp and Sante Gnerre: These authors contributed equally to this work.
Authors and Affiliations
- Broad Institute of Harvard and Massachusetts Institute of Technology (MIT), 7 Cambridge Center, Cambridge, 02142, Massachusetts, USA
Kerstin Lindblad-Toh, Manuel Garber, Or Zuk, Michael F. Lin, Pouya Kheradpour, Jason Ernst, Evan Mauceli, Lucas D. Ward, Michele Clamp, Sante Gnerre, Jessica Alföldi, Jean Chang, Federica Di Palma, Mitchell Guttman, David B. Jaffe, Marcia Lara, Jim Robinson, Xiaohui Xie, Michael C. Zody, Eric S. Lander & Manolis Kellis - Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Box 582, SE-751 23 Uppsala, Sweden
Kerstin Lindblad-Toh - MIT Computer Science and Artificial Intelligence Laboratory, 32 Vassar St. Cambridge, 02139, Masschusetts, USA
Michael F. Lin, Stefan Washietl, Pouya Kheradpour, Jason Ernst, Lucas D. Ward, Irwin Jungreis, Matthew D. Rasmussen & Manolis Kellis - Department of Biology, The Bioinformatics Centre, University of Copenhagen, DK-2200 Copenhagen, Denmark
Brian J. Parker, Ida Moltke, Jiayu Wen & Jakob S. Pedersen - EMBL-EBI, Wellcome Trust Genome Campus, Hinxton CB10 1SD, UK
Gregory Jordan, Kathryn Beal, Stephen Fitzgerald, Paul Flicek, Tim Massingham, Albert J. Vilella, Ewan Birney, Javier Herrero & Nick Goldman - Center for Biomolecular Science and Engineering, University of California, Santa Cruz, 95064, California, USA
Craig B. Lowe, Hiram Clawson, Brian J. Raney & David Haussler - Department of Developmental Biology, Stanford University, Stanford, 94305, California, USA
Craig B. Lowe - Howard Hughes Medical Institute, 4000 Jones Bridge Road, Chevy Chase, 20815, Maryland, USA
Craig B. Lowe & David Haussler - Gladstone Institutes, University of California, 1650 Owens Street, San Francisco, 94158, California, USA
Alisha K. Holloway, W. James Kent, Dennis Kostka & Katherine S. Pollard - BioTeam Inc, 7 Derosier Drive, Middleton, 01949, Massachusetts, USA
Michele Clamp - Division of Science, Research Computing, Faculty of Arts and Sciences, Harvard University, Cambridge, 02138, Massachusetts, USA
James Cuff - Department of Biological Statistics & Computational Biology, Cornell University, Ithaca, 14853, New York, USA
Melissa J. Hubisz, Andre L. Martins & Adam Siepel - Research Institute of Molecular Pathology (IMP), A-1030 Vienna, Austria
Alexander Stark - Human Genome Sequencing Center, Baylor College of Medicine, One Baylor Plaza, Houston, 77030, Texas, USA
Kim C. Worley, Christie L. Kovar, Donna M. Muzny, Richard A. Gibbs & George M. Weinstock - Genome Institute at Washington University, Washington University School of Medicine, 4444 Forest Park Blvd., Saint Louis, 63108, Missouri, USA
Wesley C. Warren, Elaine R. Mardis, George M. Weinstock & Richard K. Wilson - Genome Informatics Section, Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Elliott H. Margulies - NISC Comparative Sequencing Program, Genome Technology Branch and NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Eric D. Green - and Division of Biostatistics, Institute for Human Genetics, University of California, 1650 Owens Street, San Francisco, 94158, California, USA
Katherine S. Pollard - Department of Molecular Medicine (MOMA), Aarhus University Hospital, Skejby, DK-8200 Aarhus N, Denmark
Jakob S. Pedersen - Broad Institute of Harvard and Massachusetts Institute of Technology (MIT), 320 Charles Street, Cambridge, 02142, Massachusetts, USA
Jen Baldwin, Toby Bloom, Chee Whye Chin, Dave Heiman, Robert Nicol, Chad Nusbaum, Sarah Young & Jane Wilkinson - Human Genome Sequencing Center, Baylor College of Medicine, One Baylor Plaza, Houston, 77030, TX, USA
Andrew Cree, Huyen H. Dihn, Gerald Fowler, Shalili Jhangiani, Vandita Joshi, Sandra Lee, Lora R. Lewis, Lynne V. Nazareth, Geoffrey Okwuonu & Jireh Santibanez - Genome Institute at Washington University, Washington University School of Medicine, 4444 Forest Park Blvd, Saint Louis, 63108, Missouri, USA
Kim Delehaunty, David Dooling, Catrina Fronik, Lucinda Fulton, Bob Fulton, Tina Graves, Patrick Minx & Erica Sodergren - Research Institute of Molecular Pathology (IMP), A-1030 Vienna, Austria
Erica Sodergren
Authors
- Kerstin Lindblad-Toh
- Manuel Garber
- Or Zuk
- Michael F. Lin
- Brian J. Parker
- Stefan Washietl
- Pouya Kheradpour
- Jason Ernst
- Gregory Jordan
- Evan Mauceli
- Lucas D. Ward
- Craig B. Lowe
- Alisha K. Holloway
- Michele Clamp
- Sante Gnerre
- Jessica Alföldi
- Kathryn Beal
- Jean Chang
- Hiram Clawson
- James Cuff
- Federica Di Palma
- Stephen Fitzgerald
- Paul Flicek
- Mitchell Guttman
- Melissa J. Hubisz
- David B. Jaffe
- Irwin Jungreis
- W. James Kent
- Dennis Kostka
- Marcia Lara
- Andre L. Martins
- Tim Massingham
- Ida Moltke
- Brian J. Raney
- Matthew D. Rasmussen
- Jim Robinson
- Alexander Stark
- Albert J. Vilella
- Jiayu Wen
- Xiaohui Xie
- Michael C. Zody
- Kim C. Worley
- Christie L. Kovar
- Donna M. Muzny
- Richard A. Gibbs
- Wesley C. Warren
- Elaine R. Mardis
- George M. Weinstock
- Richard K. Wilson
- Ewan Birney
- Elliott H. Margulies
- Javier Herrero
- Eric D. Green
- David Haussler
- Adam Siepel
- Nick Goldman
- Katherine S. Pollard
- Jakob S. Pedersen
- Eric S. Lander
- Manolis Kellis
Consortia
Broad Institute Sequencing Platform and Whole Genome Assembly Team
- Jen Baldwin
- , Toby Bloom
- , Chee Whye Chin
- , Dave Heiman
- , Robert Nicol
- , Chad Nusbaum
- , Sarah Young
- & Jane Wilkinson
Baylor College of Medicine Human Genome Sequencing Center Sequencing Team
- Andrew Cree
- , Huyen H. Dihn
- , Gerald Fowler
- , Shalili Jhangiani
- , Vandita Joshi
- , Sandra Lee
- , Lora R. Lewis
- , Lynne V. Nazareth
- , Geoffrey Okwuonu
- & Jireh Santibanez
Genome Institute at Washington University
- Kim Delehaunty
- , David Dooling
- , Catrina Fronik
- , Lucinda Fulton
- , Bob Fulton
- , Tina Graves
- , Patrick Minx
- & Erica Sodergren
Contributions
K.L-.T., E.S.L. and M.K. led the project and oversaw the analysis. K.L.-T. M.C., J.Ch., E.H.M., E.D.G. and E.S.L. planned the project. K.L.-T., F.D.P., M.L., E.S.L., K.C.W., C.L.K., D.M.M., R.A.G., W.C.W., E.R.M., G.M.W. and R.K.W. oversaw or significantly contributed to data generation. S.G. assembled the 2× genomes. Major contributions to analysis were made by M.Ga., O.Z. and M.C. to evaluate measures and patterns of evolutionary selection, M.F.L. to evaluate protein-coding potential and translational readthrough, B.J.P., S.W. to analyse RNA structures and families, P.K. on regulatory motifs and motif instances, J.E. on chromatin states, G.J. on codon-specific positive selection, E.M. on promoter motif, L.D.W. on GWAS overlap with conserved elements, C.B.L. on exaptation and A.K.H., K.S.P. on HARs and PARs. J.A., K.B., H.C., J.Cu., S.F., P.F., M.Gu., M.J.H., D.B.J., I.J., W.J.K., D.K., A.L.M., T.M., I.M., B.J.R., M.D.R., J.R., A.St., A.J.V., J.W., X.X., M.C.Z., E.B., E.H.M., J.H., D.H., A.Si., N.G. and J.S.P. performed or oversaw various analyses. K.L.-T., E.S.L. and M.K. wrote the paper with input from the other authors.
Corresponding authors
Correspondence toKerstin Lindblad-Toh, Eric S. Lander or Manolis Kellis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
This file contains Supplementary Notes and Methods, Supplementary Data Sets, Supplementary References, Supplementary Tables 1-15 and Supplementary Figures 1-24 with legends. (PDF 8982 kb)
PowerPoint slides
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Lindblad-Toh, K., Garber, M., Zuk, O. et al. A high-resolution map of human evolutionary constraint using 29 mammals.Nature 478, 476–482 (2011). https://doi.org/10.1038/nature10530
- Received: 25 January 2011
- Accepted: 05 September 2011
- Published: 12 October 2011
- Issue Date: 27 October 2011
- DOI: https://doi.org/10.1038/nature10530