Immune self-reactivity triggered by drug-modified HLA-peptide repertoire (original) (raw)
Main
Abacavir (Supplementary Fig. 1) is a guanosine-related pro-drug that causes reverse transcriptase chain termination in HIV-1 infection8. T cells carrying the CD8 antigen (CD8+ T cells) from patients with resolved AHS, and from abacavir-naive, HLA-B*57:01+ healthy donors, proliferate and acquire effector functions in response to abacavir in vitro8. Moreover, abacavir-specific T cells are not activated by antigen-presenting cells expressing the closely related natural allotypes HLA-B*57:03 (Asp114Asn; Ser116Tyr), HLA-B*57:02 (Asp114Asn; Ser116Tyr; Leu156Arg) and HLA-B*58:01 (Met45Thr; Ala46Glu; Val97Arg; Val103Leu)8, suggesting that abacavir-HLA-B*57:01 specificity is particularly sensitive to the F-pocket architecture, namely residue 116. We examined the functional capacity of the natural allotype HLA-B*57:11, which differs from HLA-B*57:01 in the C/E pocket and environs (Ile94Thr; Ile95Leu; Val97Trp) to stimulate abacavir-specific T cells. Like HLA-B*57:03, HLA-B*57:02 and HLA-B*58:01 (ref. 8), HLA-B*57:11 was not functional in activating abacavir-specific T cells from HLA-B*57:01 donors (Fig. 1a), indicating that abacavir-HLA-B*57:01 specificity maps to the C-terminal end of the antigen-binding cleft (C, D, E and F pockets).
Figure 1: Specificity of abacavir T-cell responses and binding to HLA-B*57:01.
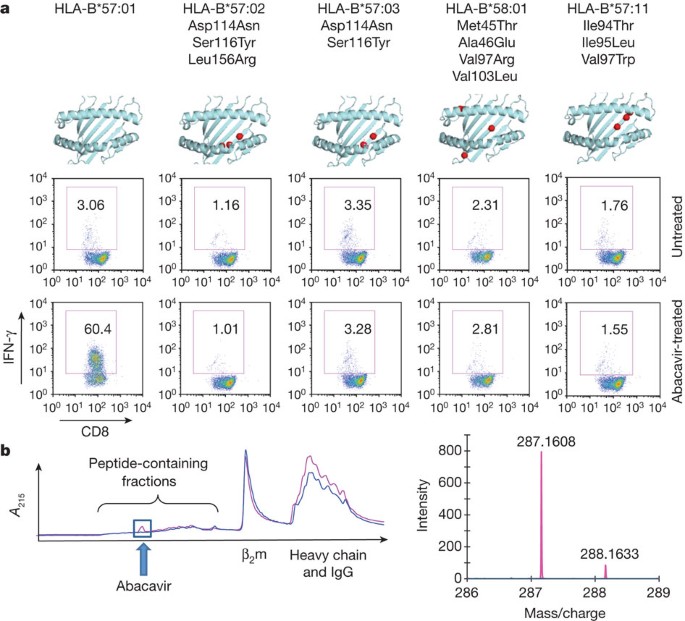
a, An abacavir-responsive T-cell line raised in vitro from an HLA-B*57:01+ donor was re-stimulated with C1R or B-LCL antigen-presenting cells expressing the indicated HLA allotypes, in the presence or absence of abacavir22. Flow plots are shown of responding T cells gated for CD3 and co-stained for CD8 and IFN-γ. Boxed areas indicate the percentage of responding T cells. Schematics show the location of HLA polymorphisms. b, Separation of peptides from HLA-B*57:01 (magenta) and HLA-B*57:03 (blue) by reverse-phase high-performance liquid chromatography after treatment of cells with abacavir. Arrow indicates the standard retention time for abacavir. β2m, β2-microglobulin. Fractions were analysed by LC–MS/MS (right). Abacavir was detected in HLA-B*57:01 preparations (mass spectra: predicted MH+ = 287.1615, Dmass (the mass difference between observed and theoretical) = 0.0007 a.m.u., 2 p.p.m. mass accuracy).
We speculated that abacavir, or a metabolite thereof, might either covalently modify a cellular protein or peptide to generate a novel immunogenic ligand or alter the peptide repertoire in some other way. Two of the most functionally important positions (114 and 116) controlling abacavir reactivity distinguish HLA-B*57:01 from HLA-B*57:03 (ref. 8), allowing us to probe how these positions alter the specificity of bound peptides and the impact of abacavir on the peptide repertoire. We therefore characterized the peptides bound to affinity-purified HLA-B*57:01 and HLA-B*57:03 molecules from untreated and abacavir-treated cell lines by mass spectrometry9. In abacavir-treated cells, reverse-phase high-performance liquid chromatography fractions from purified HLA-B*57:01, but not HLA-B*57:03, contained unmodified abacavir (Fig. 1b). Abacavir is a pro-drug, but none of its metabolites were detected by liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of the eluted material. These findings indicated that abacavir itself interacted non-covalently and specifically with HLA-B*57:01.
The repertoire of self peptides presented by HLA-B*57:01, HLA-B*57:03 and HLA-B*58:01 in the presence and absence of abacavir was examined further (Fig. 2 and Supplementary Table 1). The sequence motif characteristic of peptides that bind HLA-B*57:01 (ref. 10) was verified to contain a Ser/Thr→Ala/Val at peptide position 2 (P2) and a Trp→Phe at the C terminus of the peptide (PΩ) (Supplementary Table 1). HLA-B*57:03 had the same P2 preference as HLA-B*57:01, but the PΩ preference was reversed with Phe→Trp (Supplementary Table 1). After treatment of antigen-presenting cells with abacavir we observed a change in the nature of the peptides bound to HLA-B*57:01, but not for peptides bound to HLA-B*57:03 or HLA-B*58:01 (Fig. 2a and Supplementary Table 1). This was characterized by an increase in the number of HLA-B*57:01 ligands with non-canonical Ile or Leu at PΩ (Fig. 2a and Supplementary Table 1). In contrast, no change was detected in the preferred P2 residues selected by HLA-B*57:01 (Fig. 2a and Supplementary Table 1) or the length of peptides recovered (Fig. 2b–d). These novel peptides represented about 20–25% of the recoverable peptide repertoire, indicating a massive shift in self-antigen presentation (Fig. 2a, e), which was consistent with the 33% estimated occupancy of HLA-B*57:01 complexed with abacavir (Supplementary Table 2a). The impact of F-pocket polymorphism on abacavir recognition by T cells (Fig. 1a), the co-purification of abacavir with HLA-B*57:01-peptide complexes (Fig. 1b) and the impact of abacavir on selection of PΩ residues (Fig. 2a) led us to propose that abacavir bound specifically to the antigen-binding cleft of HLA-B*57:01. This is consistent with the dependence of abacavir presentation on TAP (transporter associated with antigen processing) and tapasin, implying a normal pathway of peptide loading8. This would result in a large proportion of the normal repertoire of peptides being sterically hindered from binding to and stabilizing HLA-B*57:01. The altered stereochemistry of the antigen-binding cleft facilitates the binding of a new repertoire of peptides containing immunogenic neo-epitopes. Consistent with this view, HLA-B*57:01 bound to peptides (Supplementary Table 2b) containing a PΩ-Trp did not further stabilize recombinant HLA-B*57:01 in the presence of abacavir. In contrast, for five of the neo-self peptides eluted specifically in the presence of abacavir, the corresponding refolded HLA-B*57:01–neo-self-peptide–abacavir complexes were approximately 5–10 °C more thermostable than the corresponding HLA-B*57:01–neo-self-peptide complexes (Supplementary Table 2b).
Figure 2: Abacavir modifies the PΩ F-pocket anchor residue preference of HLA-B*57:01 but not that of HLA-B*57:03.
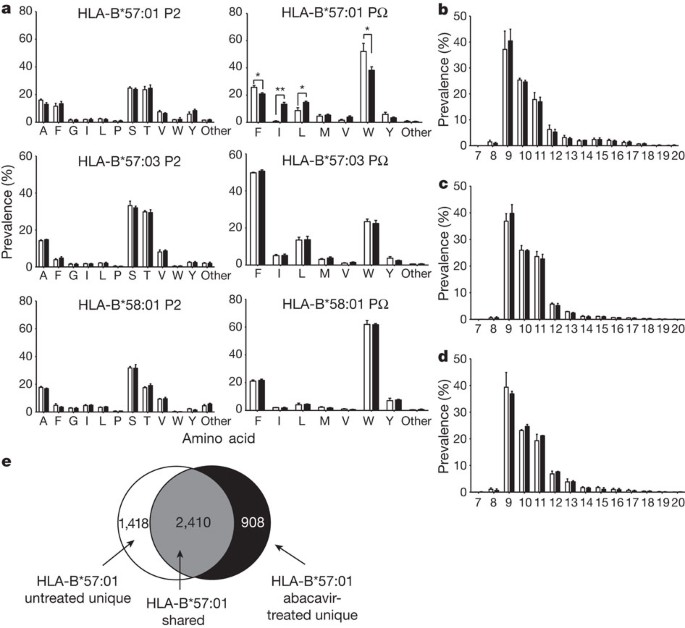
a, Frequency of amino acids occurring at P2 and PΩ in nine-residue peptides eluted from HLA-B*57:01, HLA-B*57:03 and HLA-B*58:01. Asterisk, P < 0.05; two asterisks, P < 0.001 (unpaired Student’s _t_-test). b–d, The distribution of peptide lengths from the data sets HLA-B*57:01 (b) HLA-B*57:03 (c) and HLA-B*58:01 (d). Filled and clear bars represent values from abacavir-treated and untreated cells, respectively. Values are derived from averaging prevalence across three replicate data sets of up to 3,000 distinct peptide sequences per condition. Error bars indicate s.d. across these data sets. e, The distribution of all peptides eluted from HLA-B*57:01 highlighting shared and unique peptides sequenced from untreated and abacavir-treated cells.
To test whether abacavir-specific T-cell reactivity was dependent on simultaneous presentation of abacavir and novel self peptides, we first established that T-cell-receptor (TCR) genes from a single abacavir-specific T-cell clone, transfected into Jurkat cells, specifically conferred reactivity to C1R-B*57:01 cells in the presence of abacavir (Supplementary Fig. 2a). Second, we examined the abacavir-specific T-cell response towards five neo-self peptides exclusively identified from HLA-B*57:01 complexes isolated from abacavir-treated cells. T-cell lines were raised from two different healthy HLA-B*57:01+ donors and interferon-γ (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assays were conducted using C1R-B*57:01 and T2-B*57:01 target cells with or without pretreatment with abacavir (Supplementary Fig. 2b). The addition of abacavir to T2-B*57:01 cells without the exogenous peptides did not activate T-cell clones. In contrast, the five pooled abacavir-dependent peptides added exogenously to the T2-B*57:01 cells in the presence of abacavir activated many T-cell clones (spot-forming units) in each of the two T-cell lines (Supplementary Fig. 2b). This data shows that self peptides bound to HLA-B*57:01 in the presence of abacavir specifically stimulate abacavir-responsive T cells. Thus, the selection of a novel HLA-B*57:01-peptide repertoire by abacavir exposes the normally self-tolerant T-cell compartment to previously unseen neo-self peptides resembling antigen presentation by allogeneic HLA molecules to T cells, as occurs in graft rejection11 and graft-versus-host disease12. In these circumstances a diverse repertoire of αβ TCRs may be selected by responding T cells, reflecting reactivity towards the diverse array of novel self peptides bound to HLA-B*57:01. To test this hypothesis we examined the Vβ repertoire of the TCRs selected by abacavir-specific T cells in seven unrelated HLA-B*57:01+ donors. Broadly polyclonal TCR usage (in which different T cell clones undergo expansion in response to abacavir) was observed in all donors (Fig. 3). In addition, for donors 6 and 7 the T-cell repertoire was assessed before and after stimulation with abacavir, revealing modest ‘private’ biases in immune repertoire that differed between these donors (Fig. 3; compare Vβ2 with Vβ8 in these donors before and after stimulation). Moreover, analysis of the antigen-binding complementarity-determining region 3 sequences in abacavir-specific TCRs did not reveal biased patterns (Supplementary Table 3). TCR selection was therefore consistent with the diverse array of stimulating ligands in abacavir-treated cells, with no evidence of TCR bias as frequently observed in T-cell responses to single, novel ligands13. This observation is distinct from the reported sharing of a narrow repertoire of drug-specific T cells reactive in HLA-B*15:02+ patients with carbamazepine-induced SJS or toxic epidermal necrolysis (TEN)14. The abacavir-specific, polyclonal TCR usage is consistent with the presentation of diverse neo-self peptides as a result of drug-induced ‘altered self’.
Figure 3: Vβ usage of abacavir-reactive CD8 T cells from HLA-B*57:01 positive healthy blood donors.
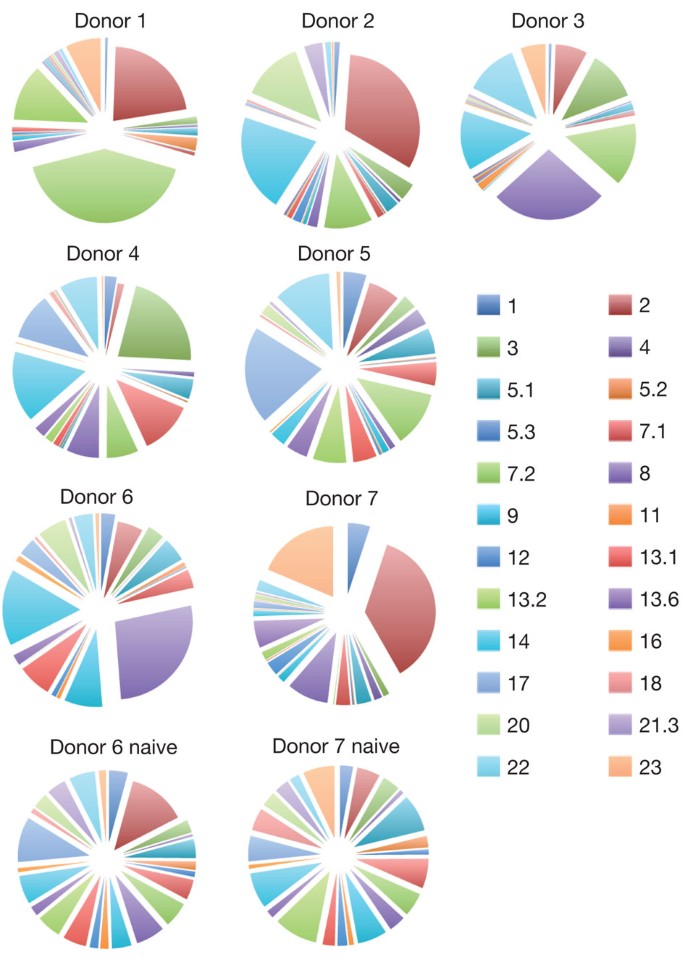
T-cell receptor Vβ usage of abacavir-reactive T cells determined by flow cytometry of in vitro activated T cells from the blood of seven HLA-B*57:01+ healthy donors. Abacavir-reactive cells were identified on the basis of IFN-γ production in response to abacavir-loaded C1R-B*57:01 and their Vβ usage is shown. The Vβ repertoire of the total CD8 T-cell population before stimulation with abacavir (naive) is shown for donors 6 and 7 for comparison.
Next we individually expressed and refolded HLA-B*57:01 in complex with two self peptides (LTTKLTNTNI, ‘LTTK’, cytochrome c oxidase subunit 2; RVAQLEQVYI, ‘RVAQ’, small nuclear ribonucleoprotein Sm D3) that had been exclusively isolated from HLA-B*57:01 in the presence of abacavir and both of which possessed the preferred PΩ residue, Ile, common in neo-peptides eluted in the presence of abacavir. Indeed, HLA-B*57:01 refolded extremely poorly with these self peptides in the absence of abacavir, which is consistent with the altered binding motif revealed by the peptide elution data. We crystallized the HLA-B*57:01–LTTK–abacavir and HLA-B*57:01–RVAQ–abacavir complexes and determined their structures to 1.9 Å and 1.6 Å resolution, respectively (Supplementary Table 4). The structure of the HLA-B*57:01–LTTK–abacavir complex was very similar to that of the HLA-B*57:01–RVAQ–abacavir complex (Supplementary Fig. 3a), revealing that the mode of abacavir binding is conserved when disparate abacavir-specific self peptides are presented by HLA-B*57:01. The HLA-B*57:01–LTTK–abacavir complex closely resembled the structure of the previously reported complex of HLA-B*57:01 with the self peptide LSSPVTKSF (ref. 8). Thus, the presence of abacavir, which was unambiguously observed to be non-covalently bound within the antigen-binding cleft (Fig. 4a, b), did not markedly alter the conformation of the antigen-binding cleft itself. Moreover, the conformation of the bound peptide, although not sitting as deep within the antigen-binding cleft as the LTTK self peptide, nevertheless adopted a slightly bulged conformation that was reminiscent of how longer major histocompatibility complex (MHC) class I peptides (more than 10 residues long)15 can bind within the antigen-binding cleft (Fig. 4b).
Figure 4: Structure of HLA-B*57:01–abacavir–peptide complex.
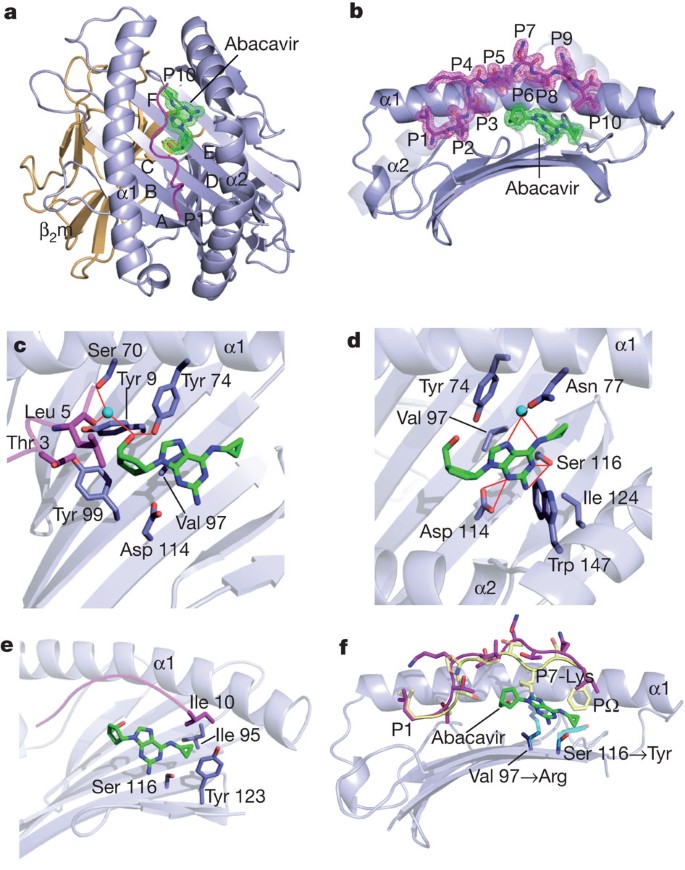
a, Abacavir within the peptide-binding groove, with the HLA pockets labelled A–F. β2m, β2-microglobulin. b, Orientation of abacavir orthogonal to a. c–e, HLA-B*57:01–LTTK–abacavir contacts cyclopentyl group (c), purine group (d) and cyclopropyl group (e). f, Superposition of the LSSPVTKSF peptide8 (yellow) on LTTK peptide (magenta) with abacavir bound to HLA-B*57:01; P7-Lys is labelled. Polymorphic differences between HLA-B*57:01, HLA-B*57:03 and HLA-B*58:01 are shown as transparent cyan sticks. Key for all panels: blue, HLA-B*57:01; orange, β2-microglobulin; magenta, peptide; green, abacavir; cyan spheres, water; red lines, hydrogen bonds; mesh, 2_F_o–F_c electron density contoured at 1_σ.
Within the HLA-B*57:01–LTTK–abacavir and HLA-B*57:01–RVAQ–abacavir complexes, abacavir was bound in an extended manner, at the base of the antigen-binding cleft, in which the plane of the aromatic rings were positioned approximately diagonally across the cleft’s β-sheet, with the cyclopentyl and purinyl moieties located in the D and E pockets, respectively, while the cyclopropyl moiety extended towards, and protruded into, the F pocket (Fig. 4c–e). The binding of abacavir did not impinge on the B pocket, which is consistent with the unchanged anchor preference at this site in abacavir-treated cells (Fig. 2a). However, the conformation of abacavir within the cleft provided a basis for understanding the global peptide repertoire shift induced by the drug, in that abacavir resided within several HLA-B*57:01 binding pockets (C, D, E and F), thereby affecting the nature of the peptides bound. For example, P7-Lys of the conventional LSSPVTKSF self peptide bound in the absence of abacavir would clash with the cyclopentyl and purinyl moieties of abacavir (Fig. 4f).
Abacavir made extensive contacts with the HLA molecule (buried surface area 450 Å2), forming numerous van der Waals contacts, five hydrogen bonds and three water-mediated hydrogen bonds, yet made few contacts with the self peptides bound within the cleft, with direct abacavir–peptide contacts being limited to P3-Thr, P5-Leu and P10-Ile for the LTTK complex and to the P10-Ile in the RVAQ complex (Fig. 4c, e). Abacavir was completely buried within the antigen-binding cleft of HLA-B*57:01–LTTK, with the peptide accounting for only 24% of the buried surface area; the HLA-B*57:01 contributed to burying the remaining 76% of abacavir’s surface area. The extensive contacts made by HLA-B*57:01 and the three moieties of abacavir (Fig. 4c–e and Supplementary Fig. 1) provided a basis for understanding why only abacavir, and not its closely related metabolites (such as carbovir), was specifically bound within the antigen-binding cleft: the abacavir metabolites are likely to have a much reduced affinity for HLA-B*57:01. Specifically, the cyclopentyl moiety of abacavir nestled against Tyr 99 (Fig. 4c), in which the O1 moiety hydrogen-bonded to Tyr 74-OH (Fig. 4d), whereas the purinyl moiety of abacavir was flanked by aromatic residues Tyr 74 and Trp 147, as well as forming van der Waals contacts with Val 97, Ser 116 and Ile 124 (Fig. 4d). Moreover, the N4 group of the purinyl moiety hydrogen-bonded to Asp 114, and the N1 and N2 groups hydrogen-bonded to Ser 116 (Fig. 4d). The cyclopropyl moiety, which resided within the F pocket, packed against Ser 116 and Tyr 123 and additionally formed van der Waals contacts with Ile 95. The presence of the cyclopropyl group within the F pocket would disfavour the presence of larger amino-acid side chains at the PΩ position (Fig. 4e), thereby providing a basis for understanding why the presence of abacavir induces a preference for smaller residues at PΩ (Fig. 4f).
The structure of the HLA-B*57:01–LTTK–abacavir complex also provided a basis for understanding the impact of HLA micropolymorphism on abacavir specificity (Fig. 4f) and our observation of abacavir co-purification only with HLA-B*57:01 (Fig. 1b). For example, abacavir cannot bind HLA-B*58:01, because the polymorphic position 97 (HLA-B*57:01 Val97Arg HLA-B*58:01) sat directly beneath, and contacted abacavir such that the presence of the long charged side chain of Arg 97 in HLA-B*58:01 would disfavour abacavir binding (Fig. 4f). Moreover, in HLA-B*57:02 and HLA-B*57:03, the replacement of Ser 116 at the base of the F pocket with the more bulky Tyr 116 residue would prevent abacavir from binding these allotypes, because the central purine group would not be accommodated (Fig. 4f). Indeed, when the HLA-B*57:01–abacavir contact sites are mapped against the HLA sequence database (http://www.ebi.ac.uk/imgt/hla/), it is evident that the constellation of contact residues is unique to HLA-B*57:01, thereby providing a basis for the exquisite specificity of abacavir towards this HLA allomorph. These findings help explain the low prevalence of AHS in African Americans who have relatively high frequencies of HLA-B*58:01, HLA-B*57:02 and HLA-B*57:03, and in whom the frequency of HLA-B*57:01 is lower than in Europe16. The penetrance of AHS in HLA-B*57:01+ individuals is about 50%, indicating that additional factors determine whether abacavir treatment induces AHS17. It is unclear whether these cofactors are genetic18 or are related to environmental factors such as drug dose. Either way, the basic mechanism involves specific co-occupancy of the HLA-B*57:01 antigen-binding cleft by abacavir and novel self peptides presenting a new ‘immunological self’ to host T cells.
Next we sought to establish the likely generality of our HLA–abacavir-associated observations, and focused on carbamazepine (CBZ) (Supplementary Fig. 4a–e). A strong HLA association is well established between HLA-B*15:02 and CBZ-induced SJS/TEN in Asian populations with odds ratios of more than 1,000 (refs 3, 4, 14). We first established that a non-covalent association existed between CBZ and HLA-B*15:02 by purifying HLA-B*15:02 peptide complexes from CBZ-treated cells (Supplementary Fig. 4). Second, sequencing of peptides bound to HLA-B*15:02 in the presence of CBZ revealed a shift in the preferred amino-acid side-chain selection with an increase in the hydrophobicity at several positions and a preference for smaller residues at the P4 and P6 positions, but no shift in anchor residue preference (P2 and PΩ) (Supplementary Fig. 4 and Supplementary Table 5a, b). The magnitude of the CBZ-induced repertoire shift, about 15%, was smaller than that observed for abacavir. This was predicted, because CBZ is likely to bind at secondary anchor sites in HLA-B*15:02, adjacent to position 156. This is based on the observation that the closely related allele, HLA-B*15:01, is not associated with CBZ-induced SJS, and a notable non-conservative difference between these two allomorphs is at position 156 (Leu, HLA-B*15:02; Trp HLA-B*15:01). The small shift in peptide repertoire might explain the restricted TCR usage against drug-specific T cells reactive in HLA-B*15:02+ patients with CBZ-induced SJS. Furthermore, automated in silico docking of CBZ into the cleft of HLA-B*15:02 predicted that CBZ binds underneath the P4/P6 residues of the peptide, adjacent to position 156 in HLA-B*15:02 (Supplementary Fig. 4c–e). Taken together, these observations for CBZ-HLA-B*15:02 peptide complexes resonated with those for abacavir–HLA-B*57:01, suggesting a general mechanism for hypersensitivity reactions towards at least two commonly used drugs with known HLA associations.
It is well established that HLA molecules can bind peptide ligands; our findings show that small-molecule drugs can specifically and non-covalently interact with defined HLA class I molecules, and subsequently alter peptide repertoire in a clinical context (Supplementary Fig. 5). These findings suggest that HLA molecules, and most probably other antigen-presenting molecules, may be surprisingly susceptible to drug modulation of antigen selection that in turn induces altered T-cell immunity. Although the observed mechanism is unlikely to account for all HLA-associated drug associations, our observations provide a potential basis for illuminating other HLA-linked drug hypersensitivities such as the very strong link between HLA-B*58:01 and allopurinol hypersensitivity syndrome19. Indeed, the extensive polymorphism of HLA molecules, the similarity of small-molecule drugs possessing ring structures (Supplementary Fig. 1) and the plethora of different drug hypersensitivities make it likely that this is a general mechanism. In this regard, the capacity of small molecular moieties to modulate immunological self could have implications for understanding the origins of autoimmunity.
Methods Summary
T-cell assays
Peripheral blood mononuclear cell samples from healthy blood donors were from the Australian Bone Marrow Donor Registry and Australian Red Cross Blood Bank. Stimulation with abacavir and cytokine assays have been described previously8.
Peptide repertoire analyses
HLA–peptide complexes were immunoaffinity purified using solid-phase-bound W6/32 monoclonal antibody20 and analysed essentially as described9.
Jurkat-transfection and activation assay
An abacavir-responsive T-cell clone (ABC12.20) was cloned into a retroviral vector (pMIG) and transduced into the Jurkat cell line. Activation was measured as the mean fluorescence intensity of staining for the early activation marker CD69.
ELISPOT assays
IFN-γ ELISPOT assays were performed using cytokine capture and detection reagents in accordance with the instructions of the manufacturer (Mabtech).
HLA-B*57:01 expression, refolding and purification
The peptides LTTKLTNTNI (cytochrome c oxidase subunit 2) and RVAQLEQVYI (small nuclear ribonucleoprotein Sm D3) were refolded with HLA-B*57:01 and abacavir and purified as described previously21.
Thermal stability assays
These were conducted on pHLA-B*57:01 samples (±abacavir) with the fluorescent dye Sypro orange in the Real Time Detection system (Corbett RotorGene 3000), to monitor protein unfolding.
In silico docking
Computational docking was performed to probe the binding of carbamazepine into HLA-B*15:02.
Crystallization and data collection
The HLA-B*57:01–LTTK–abacavir and HLA-B*57:01–RVAQ–abacavir complexes were crystallized and their structures were determined.
Online Methods
Blood samples from HLA-B*57:01+ donors
Samples from healthy blood donors were from the Australian Bone Marrow Donor Registry and Australian Red Cross Blood Bank. Institutional ethics approvals were obtained for use of all clinical material. DNA sequencing to obtain four-digit high-resolution genotyping of class I alleles at the HLA-A and B loci were performed for each donor (Victorian Transplantation and Immunogenetics Service, Victoria, Australia).
Cell culture and cell lines
Cells from the class-I-deficient lymphoblastoid cell line, C1R22,23, transfected with the relevant HLA allele were used as stimulators for functional assays and as a source of peptide for peptide elutions. Cells were maintained in RF-10 medium (RPMI (Gibco BRL) supplemented with 10% fetal calf serum (Bovogen), 7.5 mM HEPES (MP Biomedicals), 150 μg ml−1 streptomycin (Sigma), 150 U ml−1 benzylpenicillin (CSL), 2 mM l-glutamine (MP Biomedicals), 76 μM β-mercaptoethanolamine (Sigma) and 150 μM non-essential amino acids (Gibco BRL)). Treatment of cells with abacavir (Ziagen tablets; Glaxo Smith Kline) for peptide elutions was performed at 10 μg ml−1 in RF-10 for 10 days in roller bottle culture. Cells were pelleted, washed twice in PBS and snap-frozen on solid CO2.
Purification of MHC-peptide complexes
Cell pellets were ground in a Retsch Mixer Mill MM 400 under cryogenic conditions, resuspended in 0.5% IGEPAL, 50 mM Tris-HCl pH 8.0, 150 mM NaCl and protease inhibitors (Complete Protease Inhibitor Cocktail Tablet; Roche Molecular Biochemicals) at a density of 5 × 107 cells ml−1 and incubated for 1 h at 4 °C. Lysates were cleared by ultracentrifugation (180,000_g_) and HLA-peptide complexes immunoaffinity purified using solid-phase-bound W6/32 monoclonal antibody as described23. Bound complexes were eluted by acidification with 10% acetic acid. The mixture of peptides, class I heavy chain and β2-microglobulin was fractionated on a 4.6-mm internal diameter × 50-mm long monolithic C18 reverse-phase high-performance liquid chromatography column (Chromolith Speed Rod; Merck) using an ÄKTAmicro HPLC system (GE Healthcare), with a mobile phase consisting of buffer A (0.1% trifluoroacetic acid) and buffer B (80% acetonitrile/0.1% trifluoroacetic acid).
Identification of MHC-bound peptides using LC–MS/MS
Peptide-containing fractions were concentrated and loaded onto a microfluidic trap column packed with ChromXP C18-CL 3-μm particles (300 Å nominal pore size; equilibrated in 0.1% formic acid/5% acetonitrile) at 5 μl min−1 using an Eksigent NanoUltra cHiPLC system. An analytical (15 cm × 75 μm ChromXP C18-CL 3) microfluidic column was then switched in line and peptides separated by linear gradient elution of 0–80% acetonitrile over 90 min (300 nl min−1). Separated peptides were analysed with an AB SCIEX 5600 TripleTOF mass spectrometer equipped with a Nanospray III ion source and accumulating up to 30 MS/MS spectra per second. Collectively, sequence determination of up to about 3,000 peptides was conducted in three independent experiments, providing excellent technical replicates and average sampling of over 80% in each of the triplicate biological replicate experiments. Data were analysed with ProteinPilot software and peptide identities were determined subject to strict bioinformatic criteria that included the use of a decoy database to calculate the false discovery rate (FDR). A FDR cutoff of 5% was applied and the filtered data set was further analysed manually to exclude redundant peptides and known contaminants. Comparisons between data sets were performed to the following criteria: first, peptides were considered common between data sets if they appeared in at least one data set with confidence greater than the threshold for a 5% FDR for that data set, regardless of the confidence in the second data set; second, peptides were considered unique to a data set if they appeared in that data set with a confidence greater than the threshold for a 5% FDR, but did not appear in compared data set with confidence score greater than 20.
ELISPOT assays
IFN-γ ELISPOT assays were performed with cytokine capture and detection reagents in accordance with the instructions of the manufacturer (Mabtech). In brief, anti-IFN-γ antibodies were coated on the wells of a 96-well nitrocellulose plate, and duplicate wells were seeded with 60,000 target antigen-presenting cells and/or 50,000 bulk culture effector T cells per well. APCs were incubated overnight with abacavir (10 μg ml−1) and then washed three times. APCs were then incubated with peptide for 1 h at 10 μM and then washed three times. After incubation for 16 h, captured IFN-γ was detected with a biotinylated anti-IFN-γ antibody, followed by development with streptavidin horseradish peroxidase complex and chromogenic substrate. Spots were counted with an automated plate counter (AID).
Drug-specific lymphocyte culture, stimulation and Vβ characterization
Peripheral blood mononuclear cells were isolated from human whole blood or buffy coats within 24 h of collection, using Ficoll-Hypaque density centrifugation and abacavir-responsive T-cell lines established as described previously8. After 11–14 days of culture, T-cell lines were re-stimulated at a 10:1 responder:stimulator ratio with abacavir-treated (4–16 h at 10 μg ml−1 in RF-10) and irradiated (8,000 rad) C1R-B*57:01 cells that had been washed three times in RPMI. This was repeated weekly for the lifetime of the culture. After 3–4 weeks of culture, abacavir-responsive T cells were identified on re-stimulation with abacavir-treated C1R-B*57:01, using intracellular cytokine staining for IFN-γ as described previously8. Cells were phenotyped by staining with a anti-CD8 PerCP (BD Biosciences) and either anti-CD3 PE-Cy7 (eBioscience) or a panel of antibodies specific for a range of TCR Vβ (Beckman Coulter). Flow cytometry and FlowJo software (Tree Star Incorporated) were used to calculate the proportion of the abacavir-specific (CD8+/IFN-γ+) lymphocyte population expressing each Vβ.
HLA-B*57:01 expression, refolding and purification
The peptides LTTKLTNTNI and RVAQLEQVYI were synthesized by GenScript USA Inc. Mature cDNA sequences of HLA-B*57:01 and β2-microglobulin were ligated into the bacterial expression vector, pET, and recombinant protein was expressed in the BL21 strain of Escherichia coli as described previously8,21. HLA-B*57:01 (28 mg), β2-microglobulin (14 mg) and peptide (4 mg) were refolded in the presence or absence of 60 μM abacavir in 100 mM Tris-HCl pH 8.0, 0.4 M arginine, 0.5 mM oxidized glutathione, 1.5 mM reduced glutathione, 2 mM EDTA, 4 M urea, 0.2 mM phenylmethylsulphonyl fluoride in a volume of 200 ml over a 24-h period at 4 °C. The refolded protein was then dialysed for 4 h against 0.1 M urea, 10 mM Tris-HCl pH 8.0, and overnight against 10 mM Tris-HCl pH 8.0 at 4 °C using a 6–8-kDa molecular mass cut-off dialysis membrane (Spectrum). Protein was purified with fast protein liquid chromatography as described previously21.
Crystallization and data collection
The HLA-B*57:01–LTTK–abacavir and HLA-B*57:01–RVAQ–abacavir complexes were concentrated to 4 mg ml−1 in 10 mM Tris-HCl pH 8.0. Crystals were obtained at 294 K by the hanging-drop vapour-diffusion method from a solution comprising 28% PEG 8000, 0.2 M ammonium sulphate and 0.1 M cacodylate pH 6.25. The crystals grew to dimensions 0.1 mm × 0.1 mm × 0.1 mm in 4 days. Before data collection, the crystals were equilibrated in crystallization solution with 10% glycerol added as a cryoprotectant, and then flash-cooled in a stream of liquid nitrogen at 100 K. X-ray diffraction data were recorded on a Quantum-315 charge-coupled device detector at the MX2 beamline of the Australian Synchrotron. The data were integrated and scaled with MOSFLM and SCALA from the CCP4 program suite. Details of the data processing statistics are given in Supplementary Table 4.
Structure determination and refinement
The HLA-B*57:01–LTTK–abacavir structure was determined by molecular replacement as implemented in PHASER. The search model used was the structure of HLA-B*57:01 with the peptide removed (PDB accession code 2RFX). The LTTKLTNTNI peptide and a single abacavir molecule were built manually. Refinement of the model was performed in PHENIX with iterative rounds of manual building in COOT. Solvent molecules were added with COOT and the structure validated with MOLPROBITY. The final structure comprises one HLA-B*57:01–LTTK–abacavir complex in the asymmetric unit. The HLA-B*57:01–RVAQ–abacavir complex was solved using the HLA-B*57:01–LTTK–abacavir complex (minus the peptide and abacavir) as the starting point in refinement. The final refinement values are summarized in Supplementary Table 4.
Thermal stability assay
To assess the effect of the abacavir on the HLA-B*5701 stability, a thermal shift assay was performed. The fluorescent dye Sypro orange was used to monitor the protein unfolding. The thermal stability assay was performed in the Real Time Detection system (Corbett RotorGene 3000), originally designed for PCR. Each peptide was refolded at two concentrations (5 and 10 μM) in duplicate in the absence or presence of abacavir with HLA-B*57:01 in 10 mM Tris-HCl pH 8.0, 150 mM NaCl. Refolded complexes were heated from 29 °C to 90 °C at a heating rate of 1 °C min−1. The fluorescence intensity was measured with excitation at 530 nm and emission at 555 nm.
In silico docking
Computational docking was performed to probe the binding of abacavir into HLA-B*57:01 and carbamazepine into HLA-B*15:02. Two-dimensional representations of the compounds were sketched in the JMEMolecularEditor and converted to three-dimensional coordinates in ProDrg24. AutoDock Tools 1.5.4 (ref. 25) was then used to assign hydrogens, Gasteiger charges and rotatable bonds to the compounds. Each docking run was done in the absence of peptide within the binding cleft. The docking of ligands into their respective HLA peptide-binding clefts was performed with the AutoDock Vina software25. A docking grid with dimensions 42 Å × 42 Å × 42 Å, encompassing the entire peptide-binding cleft, was used.
Abacavir
The in silico docking runs with abacavir resulted in the placement of the drug with remarkable fidelity to what is observed in the crystal structure. The resultant placements of the drug are all clustered within the F pocket (Supplementary Fig. 3b). This placement was not directed, in that the entire peptide-binding groove was probed, and was not spatially constrained by the presence of peptide. The two predominant docking modes bound with docking scores (Δ_G_) of −9.5 and −8.0 kcal mol−1, which are related by an approximately 180° rotation about the central 2-aminopurine moiety. The primary placement is oriented as observed in the crystal structure, with an root mean squared deviation of 0.6 Å over all atoms, and preserves the contacts between the 2-aminopurine and cyclopropane moieties and the HLA-B*57:01 (Supplementary Fig. 3b), thereby validating the auto-docking procedure.
Carbamazepine
Because the crystal structure of HLA-B*15:02 is unavailable, it was homology modelled from the crystal structure of the closely related HLA-B*15:01 (PDB code 1XR9 (ref. 26)) (only four positions differ between these two allomorphs (HLA-B*15:01→HLA-B*15:02: Glu63Asn, Thr94Ile, Leu95Ile, His113Tyr, Trp156Leu). The computational docking of carbamazepine resulted in the exclusive clustering of the drug within the D pocket of HLA-B*15:02 (Supplementary Fig. 4c). The primary orientation bound with a docking score (Δ_G_) of −7.6 kcal mol−1. The pocket is lined with predominantly hydrophobic residues, with carbamazempine binding the side chains of residues Tyr 9, Tyr 99, Ile 66, Leu 156 and Tyr 159, the aliphatic moiety of 97 and the guanidinium group of Arg 62 (Supplementary Fig. 4d, e).
Generation and activation of the ABC12.20.Jurkat cell line
An abacavir-responsive T-cell clone was isolated by single-cell sorting of abacavir-responsive T cells identified with an IFN-γ cytokine secretion assay (Miltenyi Biotech). RNA was isolated from the outgrown clonal population, and the cDNA sequences of the TCR α and β were sequenced and identified as TRAV12-3*01-TRAJ26*01 and TRBV20-1-TRBJ2-2*01-TRBC2 using 5′-rapid amplification of cDNA ends.
Full-length cDNA ABC12.20 α and β chains were cloned into a self-cleaving 2A peptide-based MSCV–IRES–green fluorescent protein retroviral vector (pMIG)27 and transduced into the Jurkat cell line (which had been similarly transduced with genes encoding CD8αβ) using the murine stem cell virus (MSCV)-based retroviral expression system, as developed by Clontech Laboratories by means of 293T packaging cells. Cell activation was assessed after co-incubation of Jurkat cells with target APCs for 6–7 h at a 1:2 Jurkat:APC ratio in RF10. The TCR-positive Jurkat cells were identified by positive GFP fluorescence and immunofluorescence staining for CD3 and their activation was assessed as mean fluorescence intensity of the early activation marker CD69.
Accession codes
Primary accessions
Protein Data Bank
Data deposits
The atomic coordinates and structure factors for the pHLA-B*57:01–abacavir complexes are deposited in the Protein Data Bank under accession numbers 3VRJ and 3VRI.
References
- McCluskey, J. & Peh, C. A. The human leucocyte antigens and clinical medicine: an overview. Rev. Immunogenet. 1, 3–20 (1999)
CAS PubMed Google Scholar - Parham, P. & Ohta, T. Population biology of antigen presentation by MHC class I molecules. Science 272, 67–74 (1996)
Article ADS CAS PubMed Google Scholar - Bharadwaj, M. et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu. Rev. Pharmacol. Toxicol. 52, 401–431 (2012)
Article CAS PubMed Google Scholar - Chung, W. H. et al. Medical genetics: a marker for Stevens–Johnson syndrome. Nature 428, 486 (2004)
Article ADS CAS PubMed Google Scholar - Daly, A. K. et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nature Genet. 41, 816–819 (2009)
Article CAS PubMed Google Scholar - Mallal, S. et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727–732 (2002)
Article CAS PubMed Google Scholar - Hetherington, S. et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 359, 1121–1122 (2002)
Article CAS PubMed Google Scholar - Chessman, D. et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity 28, 822–832 (2008)
Article CAS PubMed Google Scholar - Purcell, A. W. et al. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J. Immunol. 166, 1016–1027 (2001)
Article CAS PubMed Google Scholar - Barber, L. D. et al. Polymorphism in the α1 helix of the HLA-B heavy chain can have an overriding influence on peptide-binding specificity. J. Immunol. 158, 1660–1669 (1997)
CAS PubMed Google Scholar - Macdonald, W. A. et al. T cell allorecognition via molecular mimicry. Immunity 31, 897–908 (2009)
Article CAS PubMed Google Scholar - Archbold, J. K., Macdonald, W. A., Burrows, S. R., Rossjohn, J. & McCluskey, J. T-cell allorecognition: a case of mistaken identity or déjà vu? Trends Immunol. 29, 220–226 (2008)
Article CAS PubMed Google Scholar - Turner, S. J., Doherty, P. C., McCluskey, J. & Rossjohn, J. Structural determinants of T-cell receptor bias in immunity. Nature Rev. Immunol. 6, 883–894 (2006)
Article CAS Google Scholar - Ko, T. M. et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens–Johnson syndrome. J. Allergy Clin. Immunol. 128, 1266–1276 (2011)
Article CAS PubMed Google Scholar - Burrows, S. R., Rossjohn, J. & McCluskey, J. Have we cut ourselves too short in mapping CTL epitopes? Trends Immunol. 27, 11–16 (2006)
Article CAS PubMed Google Scholar - Hughes, A. R. et al. Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics 5, 203–211 (2004)
Article CAS PubMed Google Scholar - Mallal, S. et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 358, 568–579 (2008)
Article PubMed Google Scholar - Martin, A. M. et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc. Natl Acad. Sci. USA 101, 4180–4185 (2004)
Article ADS CAS PubMed PubMed Central Google Scholar - Hung, S. I. et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl Acad. Sci. USA 102, 4134–4139 (2005)
Article ADS CAS PubMed PubMed Central Google Scholar - Brodsky, F. M., Bodmer, W. F. & Parham, P. Characterization of a monoclonal anti-β2-microglobulin antibody and its use in the genetic and biochemical analysis of major histocompatibility antigens. Eur. J. Immunol. 9, 536–545 (1979)
Article CAS PubMed Google Scholar - Macdonald, W. et al. Identification of a dominant self-ligand bound to three HLA B44 alleles and the preliminary crystallographic analysis of recombinant forms of each complex. FEBS Lett. 527, 27–32 (2002)
Article CAS PubMed Google Scholar - Zemmour, J., Little, A. M., Schendel, D. J. & Parham, P. The HLA-A,B ‘negative’ mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J. Immunol. 148, 1941–1948 (1992)
CAS PubMed Google Scholar - Storkus, W. J., Howell, D. N., Salter, R. D., Dawson, J. R. & Cresswell, P. NK susceptibility varies inversely with target cell class I HLA antigen expression. J. Immunol. 138, 1657–1659 (1987)
CAS PubMed Google Scholar - Schuttelkopf, A. W. & van Aalten, D. M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 (2004)
Article PubMed Google Scholar - Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010)
CAS PubMed PubMed Central Google Scholar - Roder, G. et al. Crystal structures of two peptide-HLA-B*1501 complexes; structural characterization of the HLA-B62 supertype. Acta Crystallogr. D Biol. Crystallogr. 62, 1300–1310 (2006)
Article PubMed Google Scholar - Szymczak, A. L. et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nature Biotechnol. 22, 589–594 (2004)
Article CAS Google Scholar
Acknowledgements
We thank R. Holdsworth and M. Diviney for support; N. Croft for discussions, and the staff at the MX2 beamline of the Australian synchrotron for assistance with data collection. This research was supported by a Program Grant from the National Health and Medical Research Council of Australia (NHMRC) and the Australian Research Council (ARC). S.G. is supported by a Senior Fellowship from Monash University. A.W.P. is supported by an NHMRC Senior Research Fellowship; J.R. is supported by an NHMRC Australia Fellowship.
Author information
Author notes
- Anthony W. Purcell, Jamie Rossjohn and James McCluskey: These authors contributed equally to this work.
Authors and Affiliations
- Department of Microbiology & Immunology, University of Melbourne, Parkville, Victoria 3010, Australia,
Patricia T. Illing, Lyudmila Kostenko, Zhenjun Chen, Mandvi Bharadwaj, Lars Kjer-Nielsen & James McCluskey - Department of Biochemistry and Molecular Biology and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia,
Patricia T. Illing, Nadine L. Dudek, Nicholas A. Williamson & Anthony W. Purcell - Department of Biochemistry and Molecular Biology, School of Biomedical Sciences, Monash University, Clayton, Victoria 3800, Australia,
Julian P. Vivian, Stephanie Gras & Jamie Rossjohn - Queensland Institute of Medical Research and Australian Centre for Vaccine Development, Brisbane, 4029, Queensland, Australia
John J. Miles & Scott R. Burrows - Institute of Infection and Immunity, Cardiff University School of Medicine, Heath Park, Cardiff CF14 4XN, UK ,
John J. Miles & Jamie Rossjohn - Victorian Transplantation and Immunogenetics Service, Australian Red Cross Blood Service, 100–154 Batman Street, West Melbourne, Victoria 3003, Australia ,
James McCluskey
Authors
- Patricia T. Illing
You can also search for this author inPubMed Google Scholar - Julian P. Vivian
You can also search for this author inPubMed Google Scholar - Nadine L. Dudek
You can also search for this author inPubMed Google Scholar - Lyudmila Kostenko
You can also search for this author inPubMed Google Scholar - Zhenjun Chen
You can also search for this author inPubMed Google Scholar - Mandvi Bharadwaj
You can also search for this author inPubMed Google Scholar - John J. Miles
You can also search for this author inPubMed Google Scholar - Lars Kjer-Nielsen
You can also search for this author inPubMed Google Scholar - Stephanie Gras
You can also search for this author inPubMed Google Scholar - Nicholas A. Williamson
You can also search for this author inPubMed Google Scholar - Scott R. Burrows
You can also search for this author inPubMed Google Scholar - Anthony W. Purcell
You can also search for this author inPubMed Google Scholar - Jamie Rossjohn
You can also search for this author inPubMed Google Scholar - James McCluskey
You can also search for this author inPubMed Google Scholar
Contributions
P.I. undertook functional analyses, data generation and writing of the manuscript. J.V. solved the structure and undertook structural analysis. N.D., Z.C., M.B., N.A.W., L.K., J.J.M., S.R.B., S.G. and L.K.N. contributed to data collection, experimentation and/or the provision of technical and scientific advice. A.W.P., J.McC. and J.R. are joint senior and corresponding authors—together they led the investigation, devised the project, analysed the data and wrote the manuscript.
Corresponding authors
Correspondence toAnthony W. Purcell, Jamie Rossjohn or James McCluskey.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
PowerPoint slides
Rights and permissions
About this article
Cite this article
Illing, P., Vivian, J., Dudek, N. et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire.Nature 486, 554–558 (2012). https://doi.org/10.1038/nature11147
- Received: 06 January 2012
- Accepted: 16 April 2012
- Published: 23 May 2012
- Issue Date: 28 June 2012
- DOI: https://doi.org/10.1038/nature11147